|
|
|
|
|
综述:揭示光催化剂的非共价静电工程:从分子相互作用到多场调控策略以增强电荷动力学 |
|
|
论文题目:Unlocking the non-covalent electrostatic engineering of photocatalysts: From molecular interactions to multifield tuning strategies toward enhanced charge dynamics
期刊:Advanced Powder Materials
DOI:https://doi.org/10.1016/j.apmate.2025.100338
微信链接:https://mp.weixin.qq.com/s/Zf5_jL-AXVokXYeC_t7WBg
1. 文章摘要
光催化是可持续太阳能-化学能转换中最具潜力的绿色能源技术之一。然而,光生载流子的快速复合仍然是实现高效光催化反应的关键瓶颈。近期研究进展揭示了内外部静电场在调控半导体系统电荷动力学中的关键作用。本综述重点探讨了利用非共价静电相互作用调控光催化行为的新兴策略。在材料内部,极性或铁电半导体中的自发极化可通过内置电场促进高效电荷分离,外部施加的机械应力和磁场则能通过压电效应和磁电效应进一步增强这种作用,实现对载流子传输的动态调控。除宏观领域,氢键、范德华力和π-π堆积等微观非共价静电力显著影响着表面吸附、电子结构调控和界面电荷转移过程。通过将外部场效应与半导体特性相结合,我们可以开发出稳定反应中间体、减少复合路径的创新策略,从而提升这些协同效应在能源转换和环境修复中的实用价值。本综述系统阐述了内建极化和外场在光催化系统中调控非共价静电力的机理贡献,着重分析了整合结构极性、场响应行为和界面工程的材料设计策略,以实现卓越的光催化性能。最后展望了非共价静电相互作用在光催化领域的应用前景,为理性开发更高效、更可持续的光催化系统提供理论指导。
2. 研究背景
光催化技术是目前在水处理、绿色能源生产及有机合成领域具有潜力的重要技术,受到研究者的广泛关注。在光催化过程中,半导体材料作为催化剂在适宜光照下产生载流子。这些光生载流子迁移至光催化材料表面后,可转化为高活性氧化物种(如•O2-、•OH等)或直接参与氧化还原反应。电荷分离与载流子动力学过程对光催化反应至关重要,形貌调控、表面修饰、助催化、缺陷工程及异质结构建是改善电荷分离与动力学的主要技术手段。然而这些技术存在材料合成复杂、成本高昂及量子产率低等局限,亟需从微观层面探索提升电荷传输与分离效率的新方法。
在光催化反应中施加非共价电场/磁场是一种非接触式高效技术,能显著提升光催化效率。电场与磁场可从多维度影响光催化反应过程,电场作用可改变材料的电子特性,而磁场则能调控电子自旋态。
此外,材料诱导极化对电荷分离至关重要,缺陷与非中心对称结构产生的偶极矩可形成跨材料极化场,驱动光生电荷向相反区域迁移从而实现高效分离。虽然压电、热电与铁电材料易产生极化,但其种类有限,而外电场诱导极化为电荷分离提供了更普适的解决方案。类似地,磁场作用可加速载流子运动,通过负磁阻效应、自旋极化、洛伦兹力及动生电动势等机制促进光生载流子分离。
除外场极化方法外,范德华力、π-π堆积和氢键等非共价静电力也显著影响载流子行为与光催化性能。范德华相互作用可增强电荷分离与整体光催化活性,π-π相互作用可增强光吸收与电荷转移,而氢键则主要影响表面相互作用过程。在过去的几十年里,非共价静电相互作用在光催化中的作用经历了显著发展,从早期发现逐步演进至先进光催化系统(Scheme 1和图1a)。据我们所知,尚未有综述尝试整合所有关键类型的非共价相互作用。
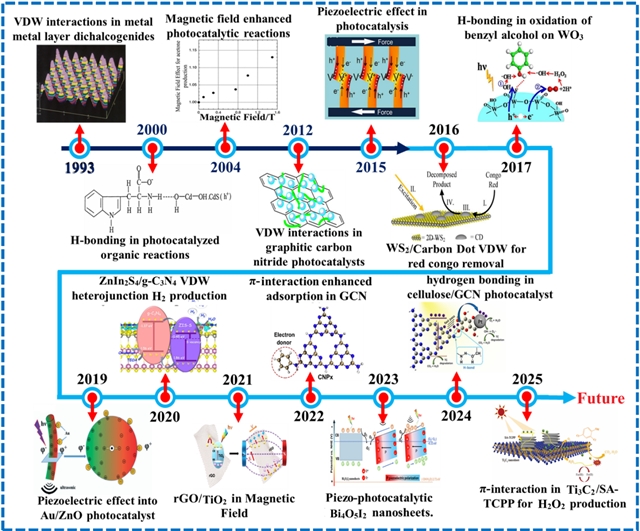
Scheme 1. 光催化中非共价静电相互作用关键发现时间轴
3. 文章概述
本综述系统论述了内场与外场对光催化剂性能的影响及其对整体光催化活性的作用机制,重点探讨了范德华力、π-π相互作用和氢键等内场作用在稳定光催化剂、促进电荷分离及强化反应物选择性吸附方面的功能。同时研究了机械应力和磁场等外场因素对光催化过程的调控作用,揭示了外场通过调制载流子行为提升光催化活性的机理。此外还探索了多极相互作用、库仑相互作用及电场效应在光催化中的应用。最后,详细讨论了非共价静电相互作用在光催化领域的应用前景与未来发展方向。
3.1 非共价静电相互作用在光催化中的关键作用
光催化材料面临诸多局限性,例如低比表面积、快速的电荷复合、有限的光吸收以及在不同介质中的分散性差等。为了克服这些挑战、提升材料性能并拓展其潜在应用,研究人员开发了多种化学修饰策略,可分为共价修饰和非共价修饰两类。图1b展示了使用共价和非共价方法对g-C3N4进行化学功能化的示意图,突出了各种修饰策略。共价修饰涉及通过氧化、羧基化、酰胺化和聚合物接枝等过程,在材料和修饰剂之间形成强化学键。另一方面,非共价修饰依赖于物理相互作用,包括范德华力、静电相互作用、π-π堆积等,以提高光催化性能,而不改变材料的核心结构。而且,非共价功能化保留了材料固有的结构和电子性质,同时能够在材料表面引入新的化学基团,增强了其在各种光催化应用中的多功能性。
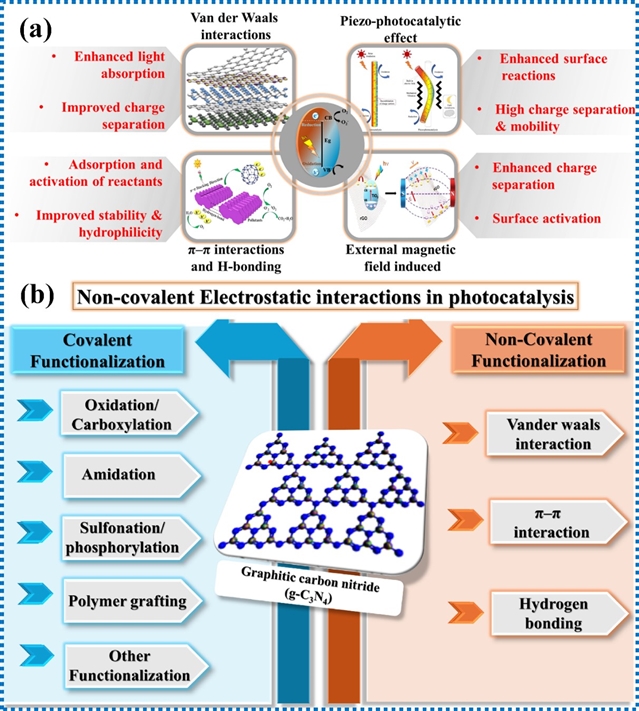
图1. (a)几种重要非共价静电相互作用对光催化效应的影响;(b)通过共价和非共价方法对g-C3N4进行化学功能化的示意图
3.2 内场调控的非共价静电相互作用
3.2.1 分子动力学中微妙的范德华力
范德华力是当两个原子或分子相互接近至外层电子云重叠时产生的弱相互作用力,偶极矩和极化率是构成范德华力的关键要素。在极性分子和中性分子中,诱导力、取向力和色散力是范德华力的主要贡献来源。范德华力高度依赖于原子间距,并随距离增大而显著减弱。尽管单个范德华力较弱,但当两个表面间存在大量范德华力共同作用时,其综合效应会变得十分显著。在光催化领域,形成异质结的两种材料间的范德华相互作用在二维层状材料中得到了广泛研究。
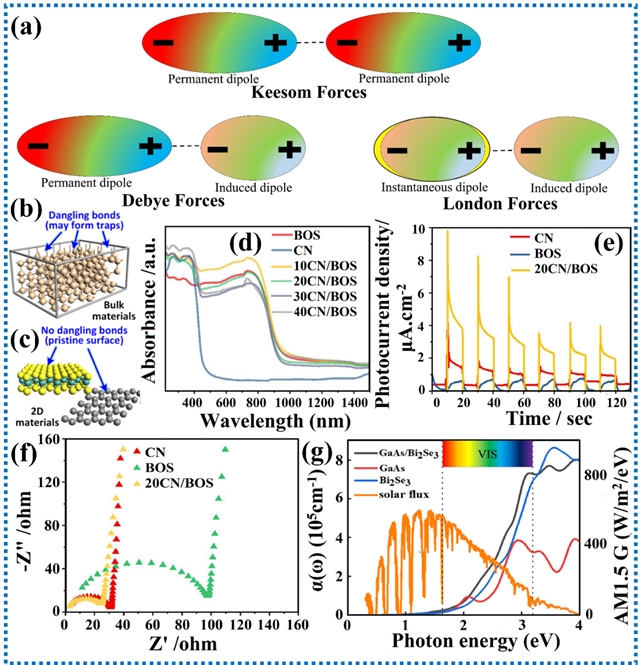
图2. 范德华相互作用的类型及其对光催化活性的影响
由于范德华异质结的相互作用能较弱,它们不会形成严格晶格匹配的异质结,而是通过规则晶格层间旋转形成纳米尺度的垂直堆叠结构。范德华异质结可通过原位生长或异位组装构建。原位法制备的异质结具有最小界面间距,能通过库仑效应和超快机制实现强组分相互作用及高效电子转移。异位法因空间位阻和能垒限制,通常导致较大界面间距、弱耦合及依赖量子隧穿的电子传输。
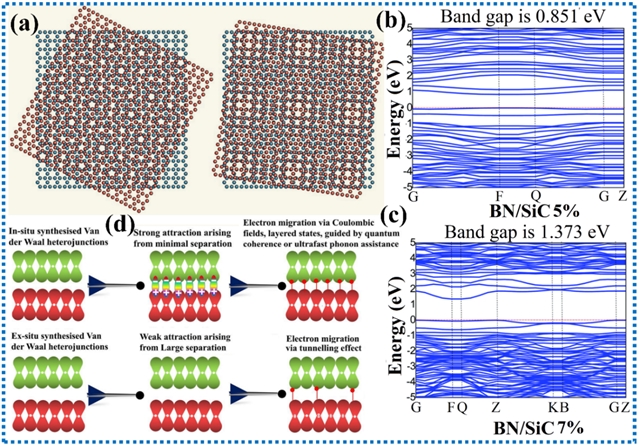
图3. 范德华异质结对能带结构和电子转移的影响
3.2.2 π相互作用的影响
具有离域π电子体系的有机光催化剂因其结构灵活性和可调控的光电性质而备受关注,这种由堆叠芳香环之间的色散力和偶极-偶极作用驱动的相互作用,显著影响材料的结晶行为、光学特性、电子性质及电荷转移过程。光催化剂中的长程π-π堆叠会导致电子-空穴对在电荷转移前快速复合,从而限制光催化效率。相反,短程π-π堆叠通过减少聚集来增强电子扩散与解离。
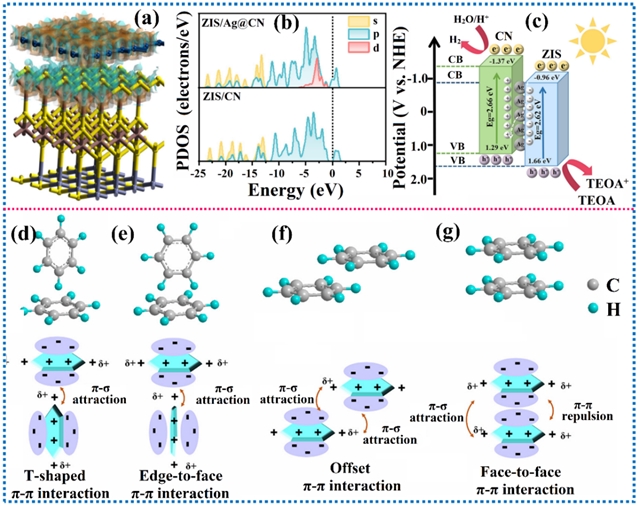
图4. 范德华异质结对光催化活性的影响以及不同类型π-π相互作用示意图
目前已构建出多种利用π-π相互作用的有机光催化材料体系,包括共价有机框架、金属有机框架、π共轭小分子、聚合物及碳基材料。早期研究聚焦于具有强光吸收和π结构的染料分子,随后拓展至导电聚合物材料。碳材料因sp2晶格、稳定性和吸附特性而受重视,MOFs因其可调控孔结构被广泛探索,g-C3N4则凭借内建电场和可见光响应特性引起关注。有机分子在氧化石墨烯上的功能化可通过共价或非共价作用实现,共价功能化会破坏延伸π共轭网络,将碳杂化由sp2转变为sp3,增加材料绝缘性并降低载流子迁移率,而非共价功能化则通过强相互作用保持π共轭体系完整。
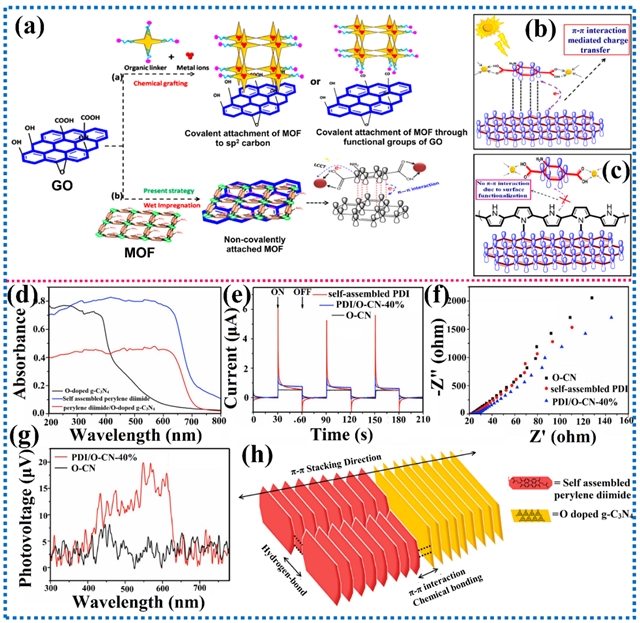
图5. π-π相互作用代表性材料体系及其对光催化活性的影响
3.2.3 氢键活化增强分子相互作用
氢键是最关键的方向性分子间相互作用之一。氢键属于非共价偶极-偶极相互作用,由氢原子在两个电负性原子间架桥形成,通常涉及N、O或F原子,在具有孤对电子的共价键供体与游离受体之间形成。氢键具有动态可逆的特性和可调节的亲和力,使其从水分子到复杂大分子体系中普遍存在。子轨道重叠与强氢键作用可增强电荷转移,提升光催化剂活性和选择性。基于这些独特性质,氢键能通过多种机制有效促进催化反应,在光催化中,氢键有助于反应物分子在光催化剂表面的吸附,其吸附强度取决于氢键类型。
图6. (a,b)氢键形成示意图;(c-l)压电材料示意图及其对电荷分布的影响
3.3 外场调控的非共价静电相互作用
施加外场辅助能在保持半导体材料本征特性的同时增强光催化活性。这些外场通常向材料内部的电子提供能量,促进其激发并提高光催化效率。此外,磁场通过超越能量传递的电子行为调控发挥着更为关键的作用,它可调节电子运动与自旋动力学,从而进一步优化光催化过程。
3.3.1 应力诱导的压电效应
为应对光生载流子的快速复合,压电光子效应已引起广泛关注,并展现出提升光催化性能的巨大潜力。近年来,压电势与极化电荷已成为增强光生载流子在光催化剂内部及表面迁移与转移的有效策略。压电效应源于机械应力与电极化的耦合作用,当具有非对称结构的介电材料受到外部机械力时,材料两侧会分别产生正负电荷。这种现象源于晶格中缺乏对称中心,导致电荷极性随施加应力方向反转而逆转。
在压电半导体中,应变诱导的内建电场会使表面能级发生偏移。正极化导致能带向下弯曲,增强电子迁移但略微降低还原电位,同时强化氧化作用;负极化引起能带向上弯曲,抑制电子转移但促进空穴传输。通过应变调控的能带弯曲程度可调节电荷转移与氧化还原电位,在光催化半导体中直接产生压电势已成为一种简捷有效的策略。部分带隙为2.5~3.5 eV的压电材料兼具光催化功能,可产生协同效应。这类材料在光照下产生光生电子-空穴对,同时应变诱导的极化电荷能增强光生载流子分离以参与氧化还原反应。
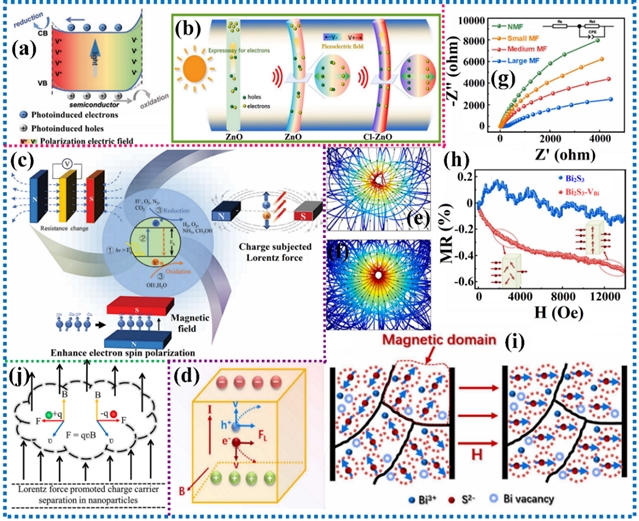
图7. (a,b)压电与光催化协同示意图及其催化机理;(c-j)磁场对电子结构和电荷分离的影响
3.3.2 磁场调控的非共价静电相互作用
随着技术进步,人类对磁现象的理解不断深化,磁场控制能力也显著提升。与其他主要提供能量的外场不同,磁场通过改变材料的电子态产生独特效应。磁场通过诱导负磁阻效应、洛伦兹力与自旋极化来增强光催化效率,这些效应可拓宽光吸收谱、促进电子-空穴分离并加速氧化还原反应,从而提升光催化性能。
3.3.3 其他非共价静电相互作用
研究人员还探索了多极相互作用、库仑相互作用及电场辅助光催化等机制。电场在光催化领域广泛应用于光电化学系统,通常通过电解池中的双电极施加,可精准控制方向与强度。在分子层面,中心金属离子周围配体产生的静电场会打破过渡金属轨道的简并性。由于电子的定向运动,外电场能在一定程度上降低载流子复合率。
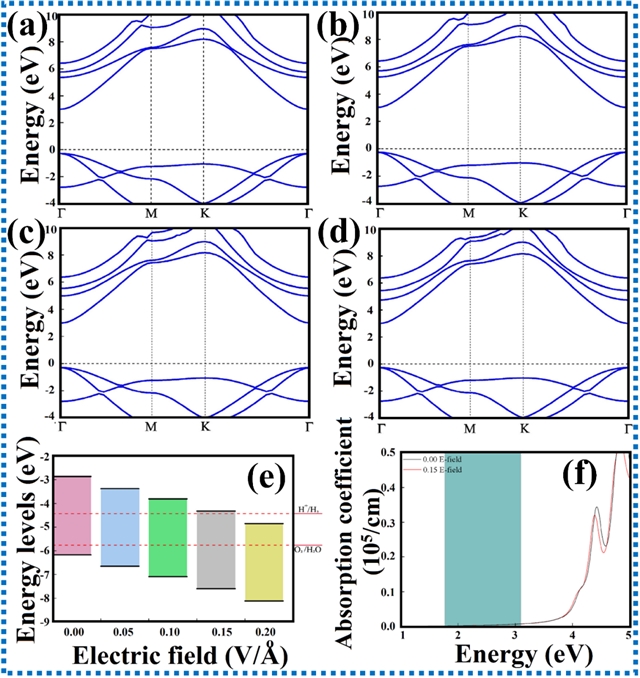
图8. 外加电场对ZnO能带结构和电荷转移的影响
多极相互作用涵盖偶极-偶极、偶极-四极及四极-四极等形式。在发光材料中,多极相互作用能调控能量转移行为,通过增强发光中心间的非辐射能量转移导致浓度猝灭,为设计高效光催化体系开辟了新路径。
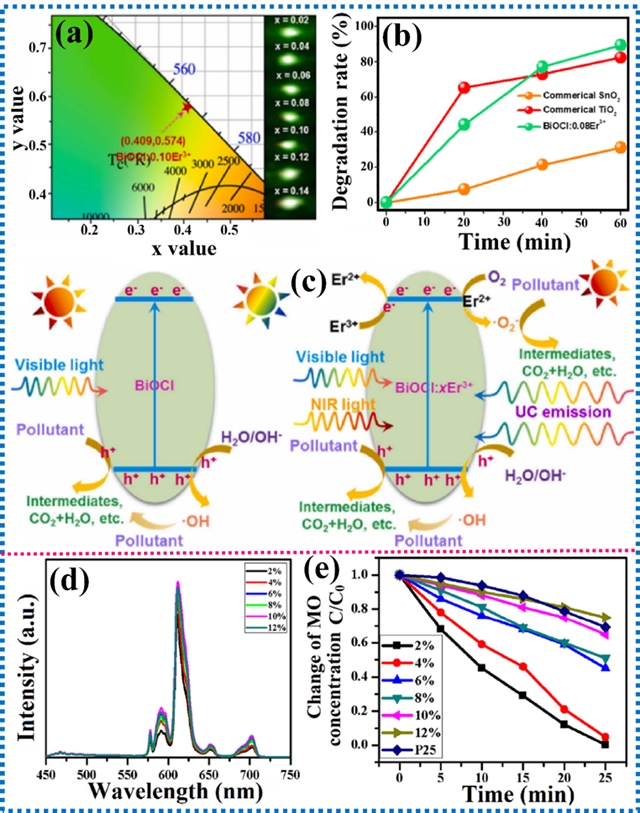
图9. 多极相互作用对光催化有机染料降解的影响
3.4 利用非共价静电相互作用实现可持续光催化
3.4.1 有机/无机反应
可见光光催化通过电子或能量转移机制进行,从底物或试剂中生成活性中间体,而非降低过渡态能量。在光照下,光催化剂进入激发态,使其能够接受或提供一个电子。这一过程通过氧化或还原淬灭循环实现光氧化还原催化。或者,激发的光催化剂可将其能量转移至底物或试剂,从而引发化学反应。非共价静电力虽弱,却是光催化中的关键作用力,可用于增强多种有机反应的光氧化还原催化性能,例如CO2还原、固氮、C-C键断裂等。范德华力、氢键、外电场磁场及压电效应等非共价静电相互作用通过增强传质过程和电荷载流子动力学来提升光催化活性,有效提高了反应速率与选择性。
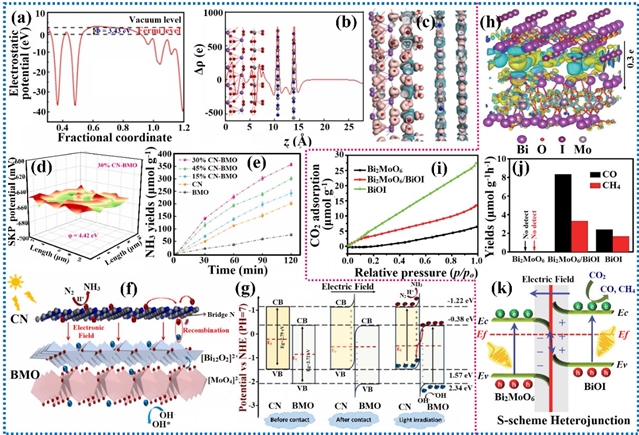
图10. 范德华异质结对电荷密度分布的影响及其在无机催化反应中的研究进展
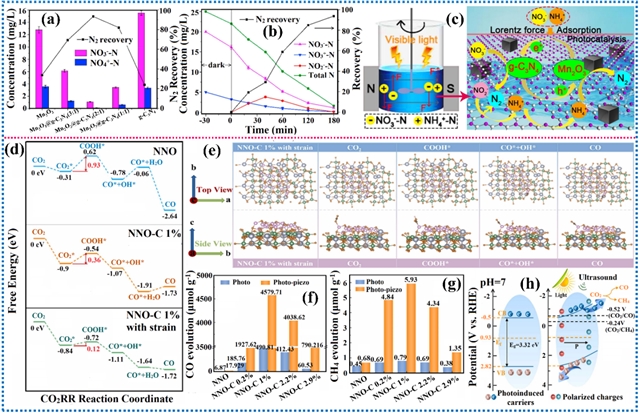
图11. 氢键、外加磁场和压电效应对电荷分布的影响及其在催化反应中的研究进展
3.4.2 能源制备
利用太阳光驱动的光催化过程生产燃料,近年来作为一种可持续且环境友好的替代方案受到广泛关注。尽管基于半导体的光催化剂已得到深入研究,但其广泛应用仍受限。主要挑战之一是其对红外线和可见光的吸收率低,显著削弱了其利用全太阳光谱的能力。此外,较低的表观量子效率也限制了其性能。突破这些限制对推动光催化技术进步、释放其在可再生能源生产方面的潜力至关重要。尽管共价修饰策略已被广泛探索用于提升光催化性能,研究人员正日益聚焦非共价相互作用,将其作为一种更精细且有效的手段来调控光催化过程。非共价相互作用能够改善电荷分离、增强表面反应活性并稳定中间体,从而影响光催化能量转换的整体效率。目前,π-π相互作用、范德华异质结、氢键、自旋极化、机械应力等非共价静电相互作用已被应用到光催化产氢中,加速了向高效绿色制氢的转型。
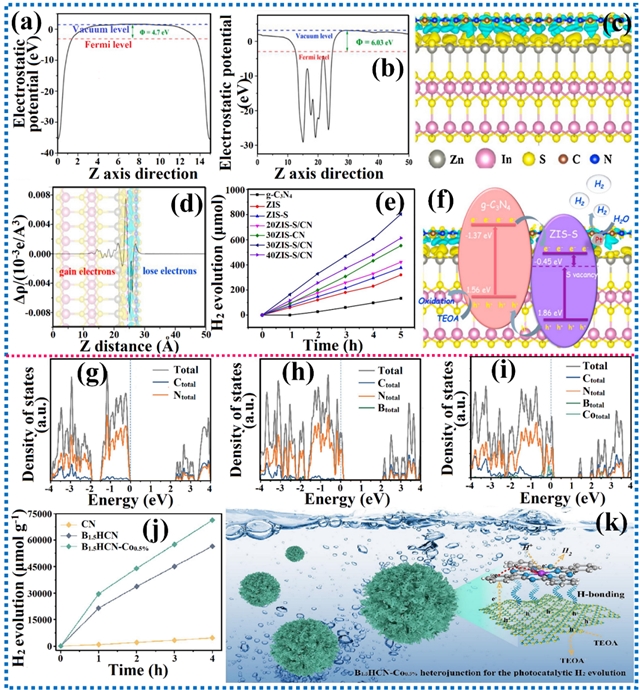
图12. 范德华异质结和氢键相互作用对电子结构和光催化产氢性能的影响
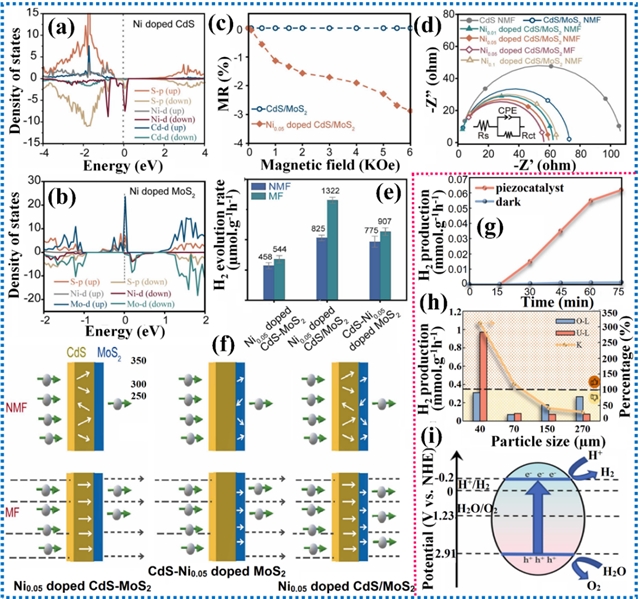
图13. 自旋极化和机械应力对电子结构和光催化产氢性能的影响
3.4.3 污染物降解
由于具有强电子耦合、可调节的电子结构以及高效的电荷分离与转移等优异特性,非共价静电相互作用近年来在污染物降解光催化反应中受到关注。利用呈现非共价静电相互作用的光催化剂,已成功降解了抗生素、染料等多种污染物。
具有互补二维材料的范德华异质结构可增强电荷分离和催化活性。合适的耦合能拓宽光吸收并改善电子-空穴分离,使范德华异质结构成为高效光催化污染物降解的一种有前景的方法。类似地,对自组装超分子有机材料的改性研究侧重于引入官能团、形成短程π-π堆积结构以及调控电子给体和受体单元,这些策略通过增强内建电场来提高电子转移速率。
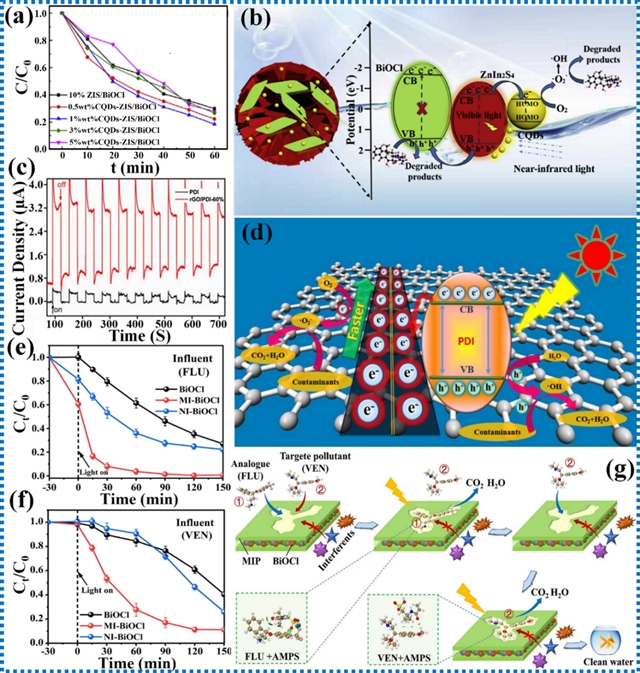
图14. 范德华力和氢键相互作用在光催化污染物降解的研究进展
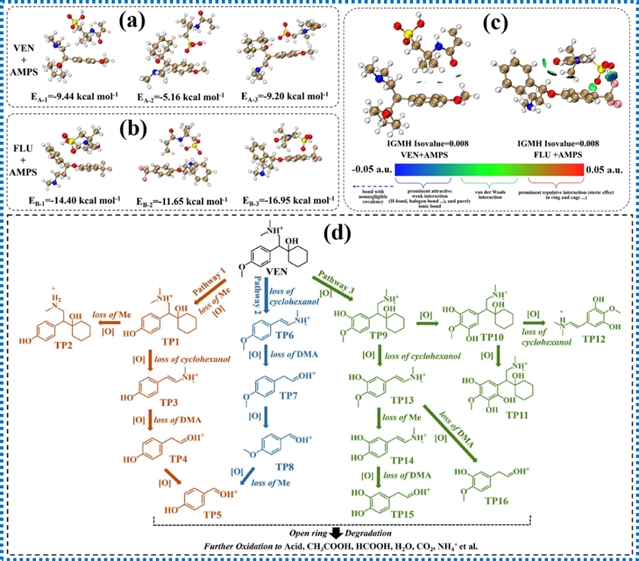
图15. 范德华力和氢键相互作用的理论计算结果及污染物降解路径
具有扩展π共轭体系的给体-受体型g-C3N4相比纯g-C3N4对双酚A的去除效率提高了2.9倍。扩展的π共轭体系不仅延长了光捕获能力,还增强了催化剂与污染物之间的π-π相互作用。这种增强促进了吸附和电子迁移,从而实现更高效的光催化降解。理论计算结果表明双酚A分子的π轨道主要位于缺电子区域,在降解过程中双酚A分子因π轨道失去电子而更易被催化剂氧化。
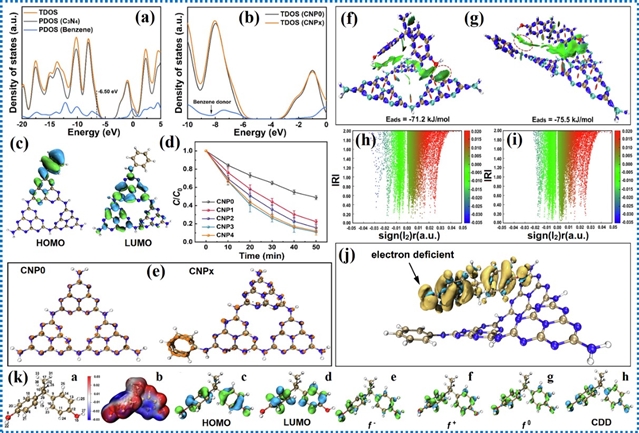
图16. π-π相互作用增强双酚A降解的理论计算结果
此外,通过将g-C3N4与生物炭复合可以提升光降解能力,优化后的材料在可见光下通过活化过硫酸盐用于四环素降解。活性提升归因于生物炭引入了吸电子基团,调控了g-C3N4的电子结构,同时增强了π-π相互作用。这种协同作用有助于抑制光生载流子的复合,从而提高光催化效率。此外,二硅酸盐的存在显著增强了g-C3N4水悬浮液中磺胺二甲嘧啶的光降解效率,主要归因于氢键相互作用。二硅酸盐中的-OH基团和桥氧(Si-O-Si)与g-C3N4的氨基以及磺胺类药物形成氢键,可溶性二硅酸盐充当了通道,促进了磺胺二甲嘧啶在g-C3N4表面的吸附和转移,从而提高了其光催化降解效率。
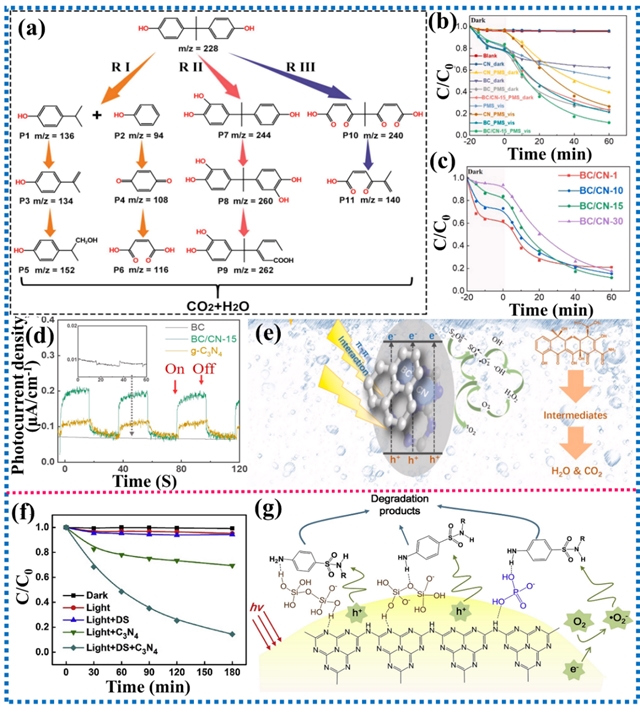
图17. π-π相互作用和氢键相互作用对光催化有机物降解的增强机制
外场辅助的光催化剂在污染物降解方面也展现出优异的光催化活性。研究人员开发了Ag2S/TiO2 Z型异质结,并研究了外磁场对四环素降解的影响。构建Ag2S/TiO2异质结增强了载流子分离并减少了复合,且外加磁场显著提高了降解率。这种光活性的增强归因于磁场诱导的自旋极化和洛伦兹力,它们促进了光生载流子的快速分离和迁移,最终以降低的活化能加速了四环素的降解。类似地,还研究了在超声振动下BaTiO3/g-C3N4复合材料对四环素的光催化降解。在超声振动下,BaTiO3和g-C3N4中产生的极化电场有助于维持异质结界面处的内建电场强度。这一过程加速了光生电荷载流子的迁移,提高了它们的分离速率,最终导致催化性能的协同增强。
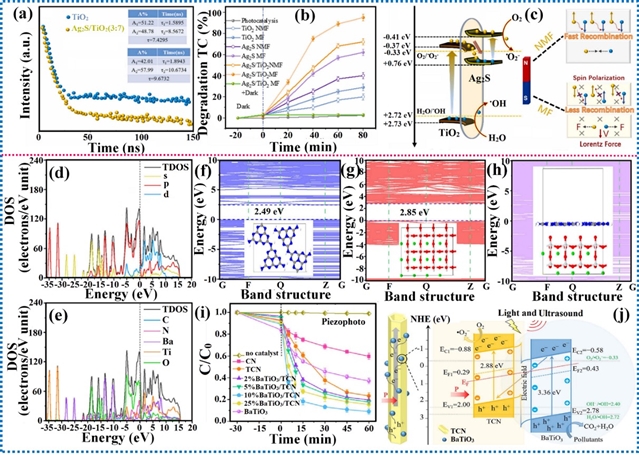
图18. 外加磁场和超声振动对光催化四环素降解的影响
4. 结论与展望
非共价静电相互作用显著改善了光催化体系的吸附、电荷转移和稳定性。范德华相互作用增强了分子和电子性质,而π-π相互作用则促进了电荷传输和电子离域。氢键作用增强了异质结中的吸附性能,减少了载流子复合,并强化了界面接触。此外,压电效应引入的内建电场促进了电荷分离,而外加磁场则进一步增强了载流子动力学和反应选择性。这些非共价静电相互作用通过改善电荷管理、增强反应物相互作用和稳定活性位点,协同优化了光催化性能,使其成为下一代光催化剂理性设计中不可或缺的要素。尽管非共价相互作用在增强光催化方面展现出巨大潜力,但仍有一些挑战和机遇需要进一步研究(Scheme 2)。
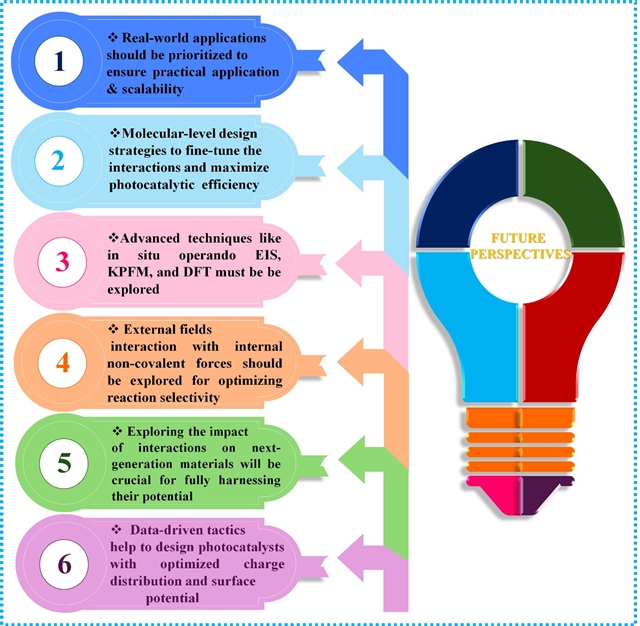
Scheme 2. 光催化中非共价静电相互作用的未来展望
(1)目前关于光催化中非共价静电相互作用的研究主要在受控实验室环境下进行,忽略了实际工业应用或环境修复中的复杂性与不可预测性。未来研究应聚焦于实际应用场景,开展现场试验与中试规模项目,弥合实验室发现与大规模应用之间的鸿沟。
(2)尽管非共价静电相互作用在光催化过程的电荷分离、表面吸附和中间体稳定化等方面发挥着关键作用,但如何精准调控其作用强度、空间构型与动态行为仍是重大挑战。未来研究应重点发展先进的分子水平设计策略,通过构筑特定表面形貌来引导定向相互作用模式。
(3)新兴的原位电化学光谱与开尔文探针力显微镜等技术为直接观测局域表面电位和实时电荷分布开辟了新途径。结合电场动态建模的先进计算方法,能够预测静电力对吸附能、过渡态及反应能量的影响机制。这些新兴方法论有望为指导下一代光催化材料的理性设计提供理论依据。
(4)密度泛函理论(DFT)与机器学习(ML)技术已成为探索和设计光催化材料的重要工具。加强数据驱动方法的研究将有助于设计出具有优化电荷分布与表面电势的光催化剂,从而实现更高效、更具针对性的性能。通过将DFT与ML相结合来预测非共价相互作用,可显著加速高效光催化体系的研发进程。
(5)需要进一步研究光催化体系内部非共价作用力与外部场之间的相互作用机制。深入理解这些相互作用关系将有助于开发调控光催化路径的新方法,从而提升量子效率、反应选择性,甚至在复杂光催化过程中实现产物分布的精准控制。
(6)新兴光催化材料(包括单原子催化剂、双原子催化剂和MXene等)所具有的独特性质可通过非共价相互作用进一步优化。通过氢键或偶极相互作用调控单/双原子催化剂的配位环境,可增强其稳定性与电子耦合效应。此外,在MXene中引入层间官能团能够改善异质结界面相容性。
引用信息:Rohit Kumar, Monika Malhotra, Anita Sudhaik, Pankaj Raizada, Xuan-Cuong Luu, Aftab Aslam Parwaz Khan, Sourbh Thakur, Tansir Ahamad, Van-Huy Nguyen, Pardeep Singh, Unlocking the non-covalent electrostatic engineering of photocatalysts: From molecular interactions to multifield tuning strategies toward enhanced charge dynamics, Adv. Powder Mater. 4(2025)100338. https://doi.org/10.1016/j.apmate.2025.100338

扫二维码 查看全文
原文链接:https://www.sciencedirect.com/science/article/pii/S2772834X25000740
特别声明:本文转载仅仅是出于传播信息的需要,并不意味着代表本网站观点或证实其内容的真实性;如其他媒体、网站或个人从本网站转载使用,须保留本网站注明的“来源”,并自负版权等法律责任;作者如果不希望被转载或者联系转载稿费等事宜,请与我们接洽。






