|
|
|
|
|
科学家开发出高通量检测单细胞mRNA动态变化的新技术scNT-seq |
|
|
细胞命运转变以及响应外界信号的过程中会改变细胞类型特异(cell-type-specific)的基因表达,而基因表达的丰度(total RNA level)是由mRNA转录、加工、降解等过程共同决定的。
在含有多种细胞类型的复杂组织和系统中,在单细胞水平准确测定这些 mRNA动态变化过程对于理解基因表达调控的重要性不言而喻。
传统的mRNA代谢标记技术利用尿苷类似物4-硫尿苷(4sU)可以有效的标记新生mRNA,但是技术上的局限性不能达到单细胞分辨率;而常规单细胞转录组测序技术虽然可以揭示不同细胞类型的稳态转录组,但是不能精确分辨特定时间里新生成的和已有的mRNA,因此用于定量研究细胞类型特异的mRNA动态调控机制十分困难。
为了应对这一挑战,美国宾夕法尼亚大学吴昊实验室开发了一种高通量检测单细胞mRNA动态变化的新方法, scNT-seq(全称single-cell metabolically labeled new RNA tagging sequencing)。
该方法创新性的整合了mRNA代谢标记 (metabolic labeling),基于液滴微流控(droplet microfluidics)的高通量单细胞转录组分析技术和最近开发的4sU化学转化反应 (chemically recode 4sU to cytosine analog);在数据分析方面,作者构建了基于unique molecular identifier (UMI) 的统计模型来更加准确地分析单细胞水平上新生成的mRNA的比例。
北京时间2020年8月31日晚23时,Nature Methods杂志在线发表了文章 “Massively parallel and time-resolved RNA sequencing in single cells with scNT-seq”。
作者应用这一技术解析了小鼠神经元快速激活过程中的单细胞基因调控网络并预测细胞状态变化轨迹,他们还进一步研究了不同胚胎干细胞状态转换过程中的mRNA动态变化的调控机制。

图1 scNT-seq的流程图。
scNT-seq具体技术流程如下:首先将4sU添加到培养基中用来标记细胞中新生成的mRNA(图1第1步)。为了在单细胞转录组中区分含有4sU标记的转录本,将4sU化学转化反应整合到液滴微流控 (Drop-seq) 流程中(图1第2-4步)。
由于捕获mRNA的barcoded beads具有区分不同转录本的UMI序列,相比较没有UMI的单细胞分析技术(e.g. scSLAM-seq),该方法不仅可以消除由于PCR扩增带来的误差,而且相对于单个mRNA分子而言能增加T-to-C的覆盖度(图1第5-8步)。
最后,利用基于UMI的二项式混合分布来准确估计每个细胞中单个基因的新生转录本。(图1第9步)
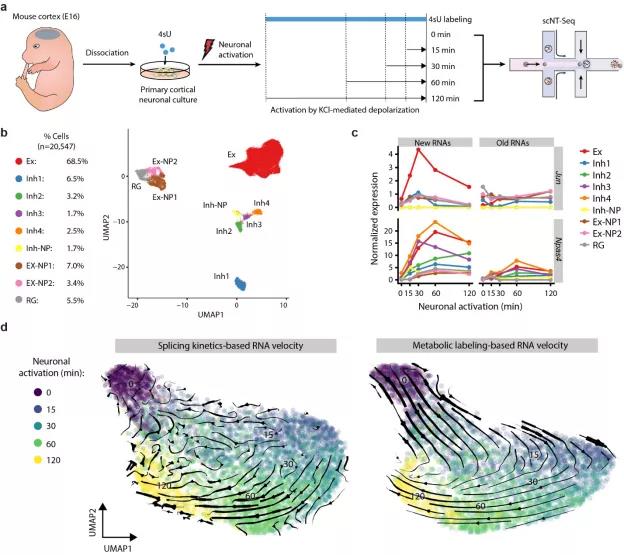
图2 利用scNT-seq技术研究小鼠大脑皮层神经元中神经活性调控的转录动态。
前人的研究表明,神经元激活后可以快速诱导(几分钟到几小时内)几百个特异基因表达,这些基因被称为神经活性调节的基因(Activity-regulated genes, ARGs)。
利用ARGs被神经活性快速诱导而在静息状态下几乎不表达的特点,可用其评估scNT-seq技术检测新生成mRNA的可靠性。作者用4sU标记原代培养的小鼠大脑皮层神经细胞,其间用氯化钾(KCl)激活这些细胞(15-120分钟)(图2a)。
作者首先将大脑皮层神经元聚类成不同细胞类型,包括兴奋性神经元(Ex)、抑制性神经元 (Inh)、神经元前体细胞(NP)等(图2b)。接下来他们分析了不同细胞类型中,ARGs基因中(如Jun和Npas4基因,图2c)新(new RNAs,产生于4sU标记的2小时内)和旧(old RNAs,产生于2小时前)转录本在不同细胞类型中的表达水平。
这些ARGs的new RNAs在不同细胞类型中有不同程度的响应,而old RNAs没有明显响应(合成于4sU标记和神经元激活前),说明scNT-seq可以准确区分新旧转录本。
在准确区分新、旧转录本的基础上,作者运用新近开发的Dynamo算法程序在兴奋性神经元中进行了基于mRNA代谢标记的RNA velocity分析(图2d右侧),能准确捕获神经元激活早期(0 – 15min)和晚期(30 – 120 min)的变化。
相较之下,传统的RNA velocity利用已拼接(无内含子)和未拼接(有内含子)的转录本信息来间接地预测细胞中基因表达的变化趋势。
由于Jun, Fos等早期响应基因内含子长度较短,基于内含子/外显子定量的传统RNA velocity方法无法准确估计这些基因的动态变化,因此无法鉴定到神经元激活早期(0 – 15min)的变化 (图2d左侧)。
而基于新生成转录本的RNA velocity能够直接利用实验准确定量的新、旧转录本的信息,推测mRNA的动态变化,不受基因结构(内含子比例)影响,兼具准确性和适用性。
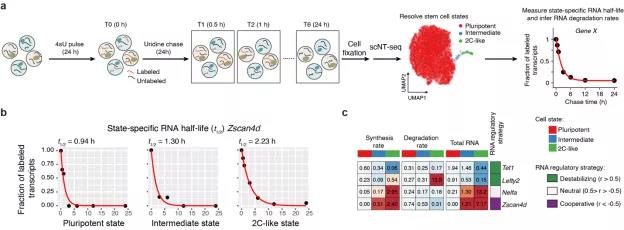
图3 利用scNT-seq技术研究小鼠胚胎干细胞不同细胞状态的mRNA丰度调节策略。
细胞中RNA的丰度由转录、加工、降解等过程共同决定,准确测定细胞群体中稀有或动态变化的细胞类型中mRNA合成和降解速率是一个重要而充满挑战的问题。
前人的研究表明,小鼠胚胎干细胞存在不同的干细胞状态,其中大约1%的细胞会自发转变为具有更高分化潜力(totipotent)的“2C-like”状态(2-cell embryos like state)。
作者通过pulse-chase的标记策略结合scNT-seq技术测定了2616个基因在不同细胞状态中的降解速率(half-life,图3a,b)。
结合4小时4sU标记实验,作者测定了445个差异表达基因的合成和降解速率。根据mRNA合成速率和降解速率在不同细胞状态间变化的趋势,这些基因可以划分为3种不同的mRNA丰度调节策略(图3c)。
有意思的是,即使采用同一种调控策略的基因,mRNA合成和降解对于不同基因mRNA丰度调节的贡献也不尽相同。例如Tet1和Lefty2基因都采用“Destabilizing”策略(mRNA合成和降解速率同向变化),但是Tet1基因主要通过降低合成速率(synthesis rate)降低2C-like细胞中mRNA丰度,而Lefty2基因主要通过提高降解速率(degradation rate)降低2C-like细胞中mRNA丰度(图3c)。
总的来说,scNT-seq是一种能在单细胞水平准确定量新生转录本的新技术。和已有的单细胞代谢标记分析方法[ref 3]相比,这项新技术兼具高准确度,高通量和低成本等优势。
因此,scNT-seq与Jay Shendure实验室的曹俊越博士新近开发的sci-fate技术,都能够广泛应用于分析大量单细胞在细胞命运决定和分化,外界信号响应,及疾病模型等动态过程中mRNA表达调控等相关的重要生物学问题。
宾夕法尼亚大学吴昊教授为通讯作者,邱琦博士和胡鹏博士为共同第一作者。邱琦博士建立了scNT-seq文库构建方法,胡鹏博士开发了scNT-seq数据分析流程,邱肖杰博士 (Whitehead Institute, https://github.com/aristoteleo) 在基于Dynamo的RNA velocity分析上提供了帮助。(来源:科学网)
相关论文信息:https://doi.org/10.1038/s41592-020-0935-4






