|
|
|
|
|
QB期刊|樊晓丹/胡杰联合发表细胞谱系追踪的方法,应用及挑战 |
|
|
论文标题:Cell lineage tracing: Methods, applications, and challenges
期刊:Quantitative Biology
作者:Shanjun Mao, Chenyang Zhang, Runjiu Chen, Shan Tang, Xiaodan Fan, Jie Hu
发表时间:28 Aug 2025
DOI:10.1002/qub2.70006
微信链接:点击此处阅读微信文章
细胞谱系追踪是解析细胞从祖细胞到分化后代发育轨迹的核心技术,堪称生命科学的 “高清导航图”—— 它不仅揭示胚胎发育、组织再生的底层逻辑,更在肿瘤异质性、耐药机制、干细胞疗法开发等临床场景中不可或缺。从早期的活体染料标记,到 Cre-loxP 遗传重组系统,再到如今的 CRISPR 条形码与单细胞测序结合,该技术已从 “追踪少数细胞” 升级为 “大规模、高分辨率、分子状态 + 谱系关系双重解析”。其核心逻辑在于实验方法(湿实验标记细胞)与计算方法(干实验重构轨迹)的协同,共同破解 “细胞从哪里来、到哪里去” 的生命谜题。
近期,Quantitative Biology 发表了香港中文大学樊晓丹教授和厦门大学胡杰教授联合团队的综述文章Cell lineage tracing: Methods, applications, andchallenges ,系统梳理细胞谱系追踪(Cell Lineage Tracing)领域的实验方法及计算方法的双体系进展,聚焦 CRISPR-Cas9 条形码、RNA velocity 等核心技术,整合跨物种基准数据集与评估体系,为发育生物学、再生医学、肿瘤学等领域提供全面技术参考,未来将通过 AI 赋能与微创标记技术突破当前瓶颈。

全文概要
细胞谱系追踪是解析细胞命运与谱系关系的关键技术,单细胞谱系追踪结合测序实现了谱系关系与分子状态的同步解析,领域已形成跨物种基准数据集,并确立了谱系树准确性等核心评估指标。该技术核心分为实验标记与计算解析体系,实验方法涵盖活体染料、Cre-loxP 等传统非 CRISPR 技术,以及条形码法、归巢 CRISPR 系统等现代 CRISPR-Cas9 技术;计算方法包括基于静态转录组的非 RNA velocity 解析法,和结合 mRNA 动力学、可预测细胞未来状态的含 RNA velocity 动态法,二者协同完成谱系轨迹的标记与重构。该技术广泛应用于发育生物学、再生医学和肿瘤学研究,可解析器官形成、干细胞稳态机制,助力组织修复疗法开发,揭示肿瘤异质性与耐药机制,同时与单细胞组学、计算生物学等深度融合,具备重要跨学科价值。目前其发展仍面临准确性不足、时间分辨率有限、多组学整合困难、可扩展性低等核心挑战,未来将聚焦技术融合、微创标记开发、AI 赋能数据处理及标准化建设,推动技术从基础研究向再生医学、个性化肿瘤治疗等临床领域转化。
细胞谱系追踪技术框架
如图1所示,细胞谱系追踪的核心是 “标记祖细胞→追踪后代的空间分布与分子特征→构建谱系树”,而单细胞谱系追踪(SCLT)通过整合单细胞 RNA 测序(scRNA-seq),实现谱系关系与基因表达状态的同步解析,可并行分析数千个细胞克隆,实现无偏倚的大规模细胞转型研究。领域内已形成覆盖秀丽隐杆线虫胚胎谱系、斑马鱼发育谱系、小鼠造血干细胞追踪、人类类器官与肿瘤模型等跨物种基准数据集,为技术验证提供金标准。衡量技术性能的三大关键指标包括谱系树准确性(与真实谱系的吻合度)、分支保真度(捕捉细胞分化分叉点的能力)与时间分辨率(动态追踪细胞状态转换的精度)。
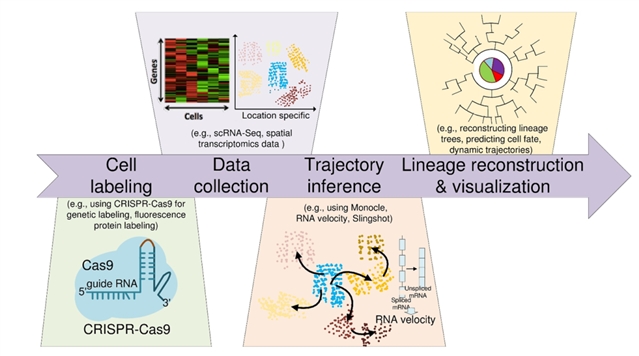
图 1. 计算驱动的细胞谱系追踪典型workflow
细胞谱系追踪核心方法
细胞谱系追踪的突破源于 “实验标记” 与 “计算解析” 的双轮驱动,两大体系持续迭代演进,形成互补协同的技术格局(图 2)。
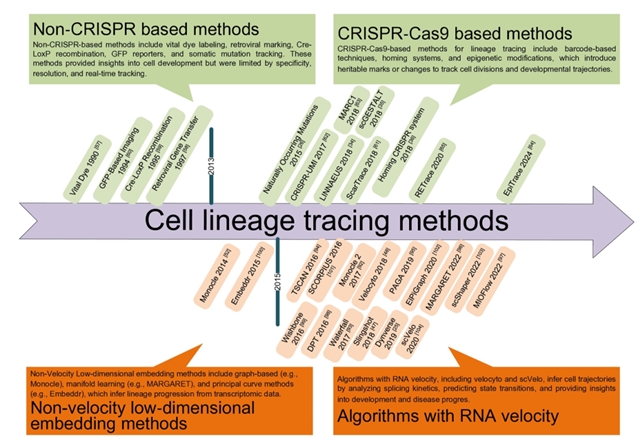
图 2. 细胞谱系追踪方法时间线
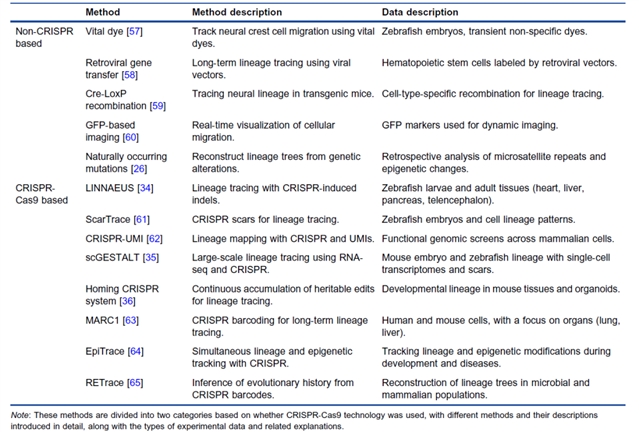
实验方法的核心是 “给细胞打独特标签”,按是否依赖 CRISPR-Cas9 分为两类(表1):传统非 CRISPR 方法包括活体染料标记、逆转录病毒介导标记、Cre-loxP 重组系统、GFP 等荧光蛋白成像及基于天然体细胞突变的回顾性追踪,这些技术奠定了谱系追踪的基础,虽存在特异性低、标记短暂或分辨率有限等局限,但在特定场景仍具应用价值;现代 CRISPR-Cas9 方法通过引入可遗传的基因条形码或突变疤痕,实现高分辨率大规模追踪,包括条形码法(如 LINNAEUS、scGESTALT)、归巢 CRISPR 系统(适配肾脏等复杂器官)与表观遗传追踪法(如 EpiTrace,微创且同步捕获表观遗传状态),部分衍生技术如 PRIME-Del、Bi-PE 进一步提升了编辑精度与效率。
表1. 实验方法汇总
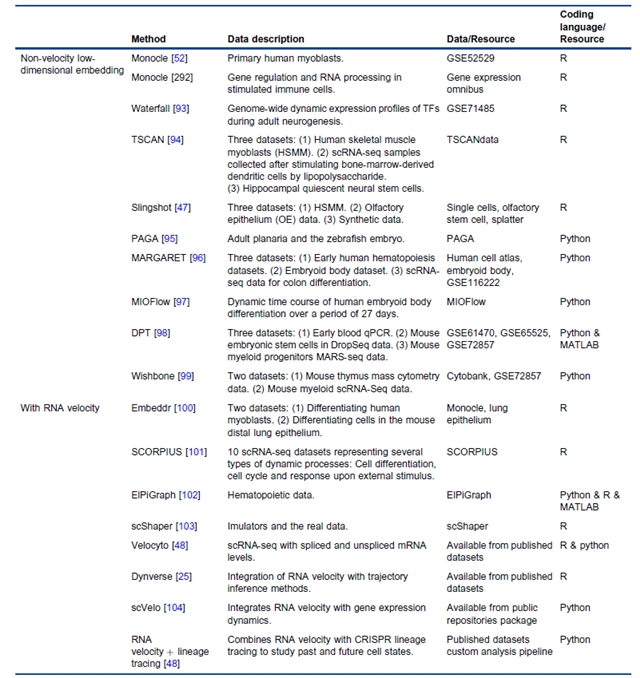
计算方法的核心是 “从海量数据中重构谱系关系”,按是否利用 RNA velocity 分为两类(表2):非 RNA velocity 方法基于单细胞转录组静态快照,通过图基方法(如 Monocle、Slingshot)、流形学习方法(如 DPT、Wishbone)、主曲线方法(如 SCORPIUS、ElPiGraph)等重构轨迹,还包括基于 SNP/CNV 的肿瘤谱系追踪工具(如 EXPANDS、MEDALT);含 RNA velocity 方法则结合未剪接 / 已剪接 mRNA 的动力学特征,预测细胞未来状态,代表工具包括 Velocyto、scVelo、PhyloVelo 等,可同时追溯细胞 “过去谱系” 与 “未来分化路径”,显著提升时间分辨率。
表2. 计算方法汇总
细胞谱系追踪方法的应用
细胞谱系追踪已成为多领域不可或缺的工具,在发育生物学中,它解析了器官形成与干细胞稳态维持机制,如揭示黑色素干细胞通过去分化维持群体稳态、CD8+ T 细胞的分化路径适应机制;在再生医学中,它助力识别组织修复关键干细胞群,为肾脏损伤修复、肝脏再生等疗法开发提供依据;在肿瘤学中,通过 CRISPR 条形码追踪肿瘤克隆动态,揭示异质性、转移进化与耐药机制,识别高转移潜能亚群与癌干细胞动态,为靶向治疗提供支撑。
该技术的跨学科价值显著,与单细胞组学结合实现 “谱系 + 多组学” 多维解析,与疾病建模联动追踪病理状态的细胞起源,与计算生物学协同突破大规模数据处理瓶颈,还为表观遗传继承、细胞异质性与进化等基础问题提供研究框架,成为连接发育生物学、再生医学、肿瘤学与计算科学的核心桥梁。
总结
本文系统整合了细胞谱系追踪领域的实验与计算双体系进展,既分类梳理了非 CRISPR/CRISPR-Cas9 介导的实验标记技术及非 RNA velocity / 含 RNA velocity 的计算重构方法,又整合了跨物种基准数据集与谱系树准确性、分支保真度等核心评估指标,全面阐述了该技术在发育生物学、再生医学、肿瘤学中的关键应用及跨学科融合价值,同时明确了当前在准确性、时间分辨率、多组学整合及可扩展性方面的核心挑战,并指明了 AI 赋能、微创标记技术开发、多模态数据融合等未来方向,为领域提供了兼具系统性与指导性的综合参考框架,助力基础研究向临床转化。
QB期刊介绍
Quantitative Biology (QB)期刊是由清华大学、北京大学、高等教育出版社联合创办的全英文学术期刊。由高等教育出版社和Wiley双平台出版和发行。QB主要刊登生物信息学、计算生物学、系统生物学、理论生物学和合成生物学的最新研究成果和前沿进展,并为生命科学与计算机、数学、物理等交叉研究领域打造一个学术水平高、可读性强、具有全球影响力的交叉学科期刊品牌。
QB期刊目前已被ESCI, PMC, Scopus, DOAJ, CSCD等国内外重要数据库收录。
《前沿》系列英文学术期刊
由教育部主管、高等教育出版社主办的《前沿》(Frontiers)系列英文学术期刊,于2006年正式创刊,以网络版和印刷版向全球发行。系列期刊包括基础科学、生命科学、工程技术和人文社会科学四个主题,是我国覆盖学科最广泛的英文学术期刊群,其中12种被SCI收录,其他也被A&HCI、Ei、MEDLINE或相应学科国际权威检索系统收录,具有一定的国际学术影响力。系列期刊采用在线优先出版方式,保证文章以最快速度发表。
中国学术前沿期刊网
http://journal.hep.com.cn

特别声明:本文转载仅仅是出于传播信息的需要,并不意味着代表本网站观点或证实其内容的真实性;如其他媒体、网站或个人从本网站转载使用,须保留本网站注明的“来源”,并自负版权等法律责任;作者如果不希望被转载或者联系转载稿费等事宜,请与我们接洽。