|
|
|
|
|
基于新分子描述符的氢键相互作用自由能预测方法|MDPI Liquids |
|
|
论文标题:Prediction of Hydrogen-Bonding Interaction Free Energies with Two New Molecular Descriptors
论文链接:https://www.mdpi.com/2673-8015/5/2/12
期刊名:Liquids
期刊主页:https://www.mdpi.com/journal/liquids
氢键作为化学与生物系统中关键的分子间作用力,其相互作用自由能的精准量化对理解溶剂化行为、优化热力学模型至关重要。然而,传统的线性溶剂化能关系(LSER)模型存在依赖实验数据、处理自缔合体系时酸 - 碱作用描述不一致等局限。为此,本文提出一种结合量子化学(QC)计算与 LSER 的新方法,通过构建两个分子描述符实现氢键自由能的简单预测,为相关研究提供了更灵活的工具。
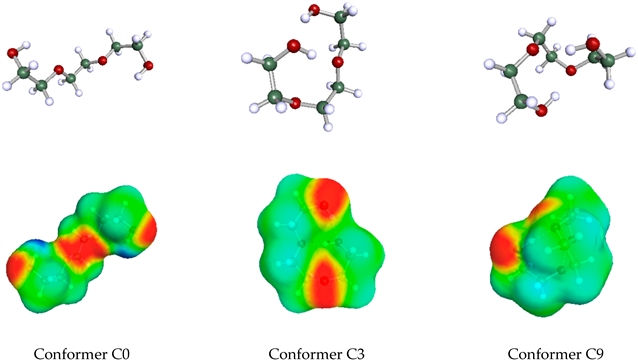
图例:三乙二醇三种构象异构体的 COSMO 几何结构(上)和 COSMO 表面(下)。下图中酸性位点用深蓝色标记,碱性位点用深红色标记。
核心方法:新分子描述符与预测公式
研究引入质子供体能力(α_G)和质子受体能力(β_G)两个描述符,分别表征分子的酸性和碱性特征。它们基于量子化学计算得到的分子表面电荷分布(σ-剖面),结合“可用性分数”(f_GA、f_GB)推导而来——可用性分数反映分子中实际参与氢键作用的酸性 / 碱性位点比例,同系物具有相近值
对于两个分子(1 和 2)的氢键相互作用自由能,计算公式为:

其中,5.71 为 25 °C时的通用常数 [((ln10)) RT],适用于单一酸性或碱性位点的分子;对于含多个远距离位点的复杂分子(如三甘醇),则需为其作为溶质和溶剂分别设置描述符。
验证与关键发现
通过与 Abraham LSER 模型对比,该方法在多数体系中表现优异:甲醇、乙醇等简单溶剂中,预测值与 LSER 估计值偏差多小于2 kJ/mol;对于三甘醇等多位点分子,使用溶剂特异性描述符后,平均偏差从8.99 kJ/mol 降至0.08 kJ/mol。
研究还报道了常见分子的描述符数据(如甲醇 α_G=0.75、β_G=1.55;水 α_G=1.56、β_G=1.61),为实际应用提供参考。
意义与局限
该方法突破了LSER对实验数据的依赖,可扩展至更多未被覆盖的体系,且在自缔合计算中满足 ,克服了传统模型的矛盾。但水体系预测偏差较大,分子构象变化对结果的影响仍需深入研究,这些将是未来优化的方向。综上,该研究为氢键相互作用的量化提供了新途径,有望推动溶剂化研究、分子热力学模型开发等领域的发展。
,克服了传统模型的矛盾。但水体系预测偏差较大,分子构象变化对结果的影响仍需深入研究,这些将是未来优化的方向。综上,该研究为氢键相互作用的量化提供了新途径,有望推动溶剂化研究、分子热力学模型开发等领域的发展。
特别声明:本文转载仅仅是出于传播信息的需要,并不意味着代表本网站观点或证实其内容的真实性;如其他媒体、网站或个人从本网站转载使用,须保留本网站注明的“来源”,并自负版权等法律责任;作者如果不希望被转载或者联系转载稿费等事宜,请与我们接洽。






