|
|
|
|
|
氮触发钌/钛氧氮化物肖特基催化剂的等离激元双功能性以实现从肼-海水中高效制氢 |
|
|
论文题目:Nitrogen-triggered plasmonic bifunctionality in ruthenium/titanium oxynitride Schottky catalyst for energized hydrazine-seawater hydrogen production
期刊:Advanced Powder Materials
DOI:https://doi.org/10.1016/j.apmate.2025.100384
微信链接:https://mp.weixin.qq.com/s/qq1oMfmo7gZkNCPg790ueA
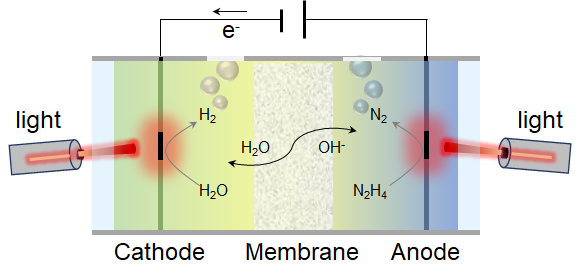
一、文章摘要
等离激元耦合肼-海水电解作为一种可提升产物效率和可再生能源利用率的先进制氢技术而备受关注。尽管将等离激元效应与双功能催化剂结合有望革新系统设计,但实现等离激元双功能催化剂的设计仍面临巨大挑战。本研究通过引入氮作为“分子开关”,成功触发钌/钛氧氮化物(Ru/TiNO0.6)的等离激元双功能性,取得等离激元增强析氢反应(过电位:13.5 mV)和肼氧化反应(过电位:222.3 mV)活性的突破性进展。该等离激元双电极系统表现出显著的性能提升:在0.2 V电压下电流密度提升34.6%(127.5→171.6 mA cm-2),同时保持接近100%的H2/N2选择性转化。通过先进表征与理论分析,我们揭示了氮元素的三重作用机制:缩小基底TiNO0.6带隙并增强光电与光热效应;强化莫特-肖特基效应生成亚稳态非晶态钌;诱导界面电荷极化并形成内建电场,协同降低催化反应能垒。这些协同效应最终实现了最优氢吸附能(ΔG*H),促进*N2H3中间体形成并改变速率决定步骤,为等离激元驱动双功能电催化建立了新的研究范式。
二、研究背景
近年来,随着全球对清洁能源需求的日益增长,氢能作为理想的绿色能源载体备受关注。传统的电解水制氢技术因析氧反应(OER)的高过电位(>1.23 V)而产生巨大能耗,严重制约其实际应用。肼介导的杂化(海)水电解技术(整体肼分解,OHzS)通过热力学更有利的肼氧化反应(HzOR,-0.33 V vs. RHE)替代OER,显著降低能耗,并避免产生爆炸性氢氧混合物,仅生成无害的氮气副产物。然而,OHzS的实际应用仍面临HER和HzOR反应路径中中间体吸附能不兼容导致的动力学瓶颈。虽然双功能催化剂可简化系统结构,但其设计需协调两种极性相反的反应路径,存在根本性挑战。
等离激元激发技术的引入为解决这一难题提供了突破性方案。通过利用光子作为非平衡能量载体,局域表面等离激元可产生电磁场和热载流子,从而绕开经典的Sabatier限制。钌基电催化剂因其兼具类铂/铱的HER活性和优异的HzOR活性,且成本低,成为肼辅助制氢的理想材料。然而,钌表面NxHy/H中间体的过强吸附会导致HzOR/HER转换频率(TOF)的动力学滞后,而其本征宽带隙光电惰性限制了其在光电催化场景的应用。通过与光吸收载体(如钛基材料)构建异质结,可同步调控原子/电子结构及光电转换能力。传统的TiO2因~3.0 eV的宽禁带、低电导率和弱金属-载体相互作用而应用受限。氮掺杂可通过能带重构、d带中心调控和界面耦合效应突破这些局限,但以往研究尚未充分利用氮掺杂,精准同步调控Ru活性位点的原子结构和钛基载体的光电特性,以建立统一的光激发双功能系统。
本研究首次在钌-二氧化钛体系中通过氮掺杂实现了等离激元双功能性。所制备的钌-钛氧氮化物在10 mA cm-2电流密度下,析氢和肼氧化过电位分别仅为34.6 mV和237.3 mV,转换频率较商业铂碳提升三倍以上。在近红外光照射下,过电位进一步显著降低。当应用于双电极电解体系时,在0.2 V电压下近红外光使电流密度从127.5提升至171.6 mA cm-2,且稳定性显著增强。通过系统表征与理论计算,研究揭示了氮元素的三重调控机制:增强光电-光热效应、诱导形成亚稳态非晶态钌、形成内建电场。这些效应协同优化了氢吸附能,促进关键中间体形成,最终建立了等离激元驱动双功能电催化的新范式。
三、创新点
1.氮作为“分子开关”触发等离激元双功能性:首次通过氮掺杂同步调控TiNO0.6载体的光吸收特性与Ru活性位点的原子/电子结构,实现光热/光电子效应协同增强的双功能催化。
2.非晶Ru/TiNO0.6肖特基结设计:氮诱导形成亚稳态非晶Ru物种,强化金属-载体相互作用,构建内建电场以优化界面电荷传输与中间体吸附能。
3.等离激元耦合肼-海水电解系统:在近红外光驱动下,实现电流密度显著提升与高稳定性运行,为低能耗制氢技术提供新路径。
四、文章概述
1. 材料合成与结构表征
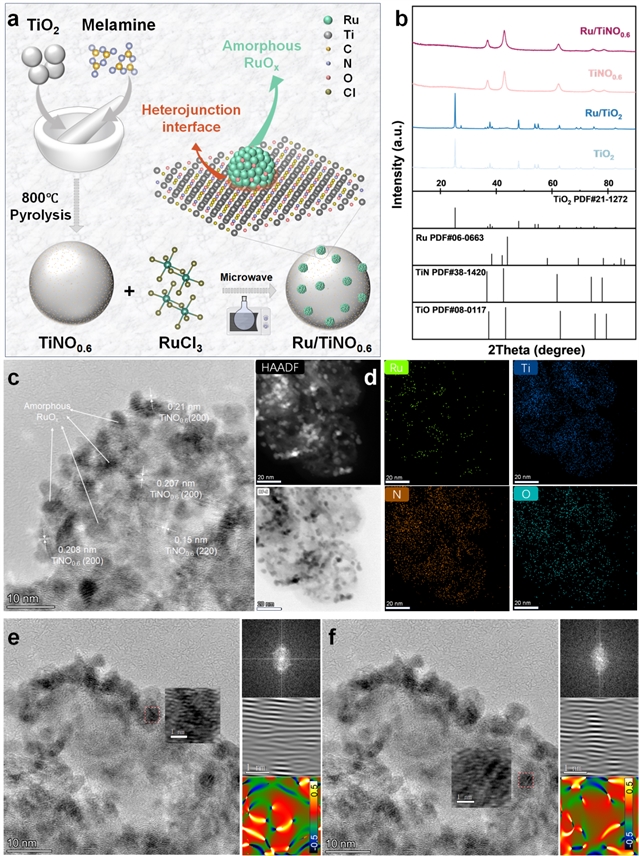
图1. 材料合成与结构表征。Ru/TiNO0.6的(a) 制备过程示意图,(b) XRD图像,(c) HRTEM图像。(d) HAADF-STEM图像和EDS元素分布图,(e, f) FFT和IFFT图像,以及插图中εyy的应变分布。
本研究首先通过高温热解将氮掺入TiO2载体,随后通过微波辅助法负载钌形成异质结。XRD图谱(图1b)显示,氮掺杂使载体从单斜相转变为面心立方结构,且未观察到明显的Ru/RuOx衍射峰,表明形成了非晶Ru。HRTEM图像(图1c)显示Ru原子呈无序排列,形成平均尺寸约4.1 nm的非晶纳米颗粒。HAADF-STEM和EDS mapping(图1d)证实Ti、N、O元素均匀分布,Ru物种负载于表面。应变分析(图1e, f)显示非晶Ru域内存在位错和异质应变,这种结构富含不饱和配位点和悬挂键,有利于催化活性提升。
2. 电子结构与化学键合分析
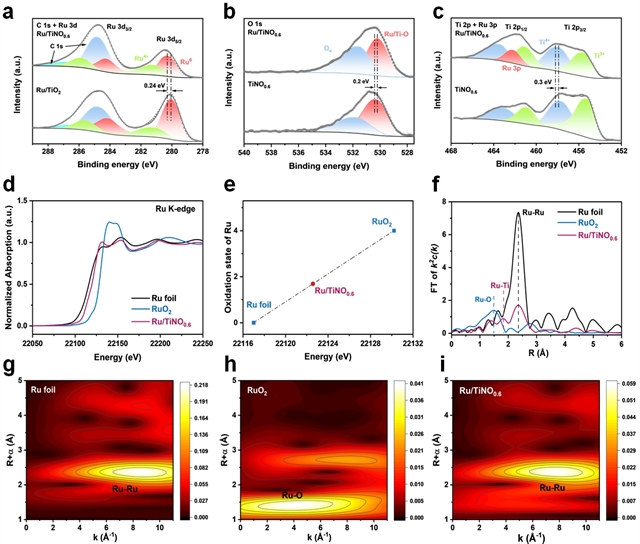
图2. 电子与成键结构表征(a) Ru 3d + C 1s ,(b) O 1s ,(c) Ru 3p + Ti 2p 高分辨XPS光谱。(d) Ru K边XANES光谱。(e) Ru/TiNO0.6中Ru的化学价态。Ru/TiNO0.6、RuO2和Ru箔样品的(f) FT谱图,(g-i) Ru K边的k3权重EXAFS谱的WT图。
XPS分析显示(图2a-c),Ru/TiNO0.6中Ru 3d峰相较于Ru/TiO2发生0.24 eV正移,表明界面存在强电子耦合。同时,与原始RuOx相比,Ru 3d峰发生负移,证实电子从TiNO基底向Ru的转移。Ru K-edge XANES光谱(图2d)显示Ru/TiNO0.6的价态介于Ru箔和RuO2之间,平均价态为+1.7(图2e)。EXAFS分析(图2f)显示存在Ru-O(~1.40 Å)、Ru-Ti(~1.84 Å)和Ru-Ru(~2.36 Å)键。小波变换分析(图2g-i)进一步证实了Ru-Ru和Ru-O键的贡献,表明非晶RuOx通过Ru-O-Ti和Ru-Ti界面键与TiNO0.6基底有效耦合。
3. 三电极体系电催化性能
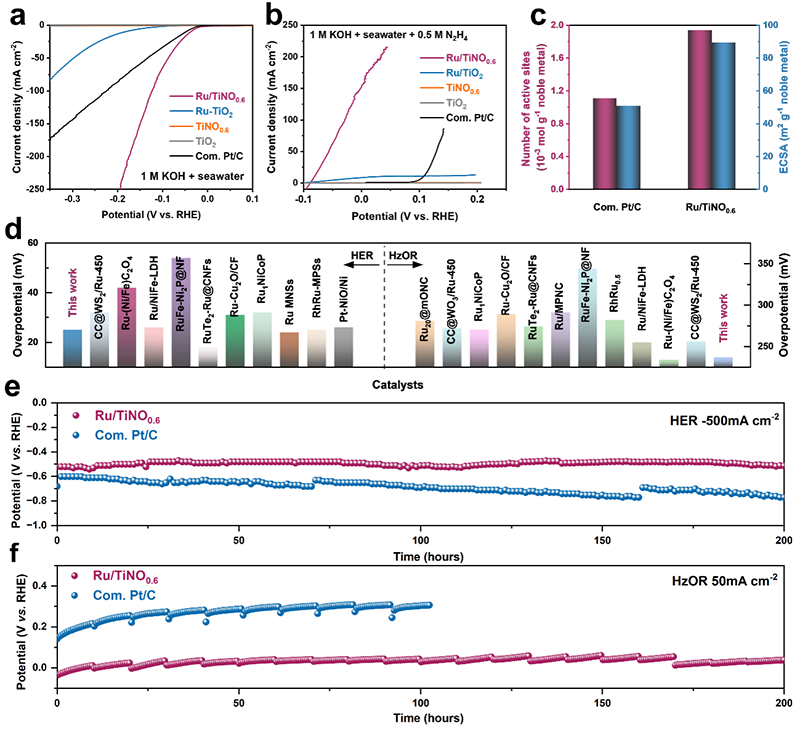
图3. 三电极体系性能表征。(a) 析氢反应在1.0 M KOH+海水溶液及(b) 肼氧化反应在1.0 M KOH+海水+0.5 M N2H4溶液中的极化曲线。(c) 通过铜欠电位沉积法测算的活性位点数量与电化学活性面积。(d) Ru/TiNO0.6与已报道贵金属基电催化剂的双功能电化学性能对比。(e) 1 M KOH+海水溶液及(f) 1.0 M KOH+海水+0.5 M N2H4溶液中Ru/TiNO0.6与商用Pt/C的计时电位测试对比。
在碱性海水环境中,Ru/TiNO0.6表现出优异的双功能催化性能(图3a-b)。HER在10/100 mA cm-2的过电位分别为45/143 mV,HzOR在10/50 mA cm-2的过电位为249.3/274 mV,显著优于商用Pt/C催化剂。Cu-UPD分析(图3c)显示Ru/TiNO0.6的活性位点数量是Pt/C的1.75倍,ECSA高达89.4 m2 g-1。与其他贵金属基电催化剂的性能对比(图3d)表明其处于领先水平。稳定性测试(图3e-f)显示,在200小时HER运行和HzOR运行中均保持优异稳定性,且性能衰减远小于商用Pt/C。
4. 近红外光耦合电化学性能
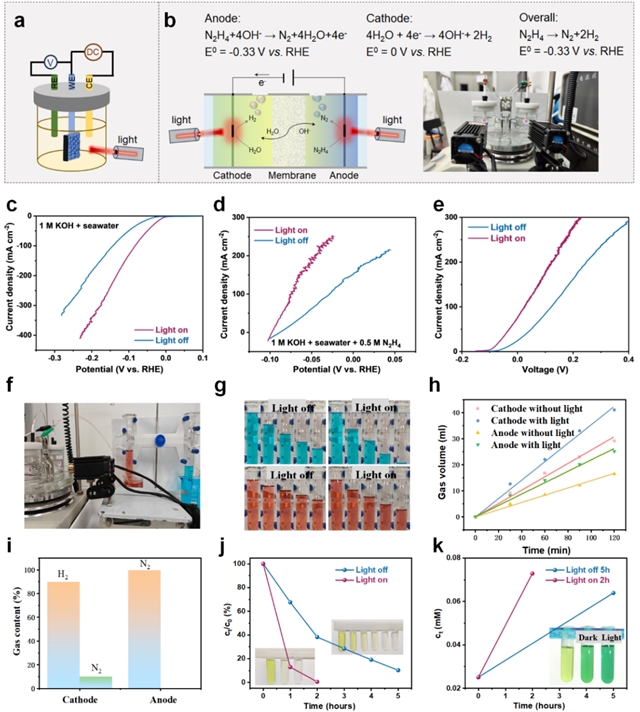
图4. 近红外光耦合电化学性能。近红外光耦合(a) 三电极测试示意图,(b) 双电极OHzS电解槽示意图。在碱性海水溶液中,光照(808 nm)开关条件下Ru/TiNO0.6的iR补偿(c) HER极化曲线和(d) HzOR极化曲线。(e) 在施加偏压和光照开启/关闭条件下,以Ru/TiNO0.6作为双功能电极在海水溶液中进行碱性OHzS的iR补偿LSV曲线。(f) 近红外光辅助双电极OHzS气体收集装置照片。(g) 在光照关闭和开启条件下(iR补偿电位:0.5 V),阴极(蓝色)和阳极(红色)收集的气体量和(h) 体积随时间变化的曲线。(i) 阴极和阳极气体含量的百分比。光照关闭和开启条件下(j) 肼分解百分比和(k) 氨生成速率随时间变化。
在近红外光照射下(图4a-b),HER过电位进一步降低至13.5 mV@10 mA cm-2(图4c),HzOR过电位降低15 mV@10 mA cm-2(图4d)。双电极OHzS系统在0.2 V下,电流密度从127.5提升至171.6 mA cm-2(图4e)。气体收集实验(图4f-h)显示,光照下阴极H2产率提升1.38倍,阳极N2产率提升1.59倍,H2/N2选择性均保持近100%(图4i)。同时,肼分解动力学加速300%,2小时内转化率达97%,且有效抑制NH3的生成至痕量水平(图4j-k)。
5.光热与光电子效应量化
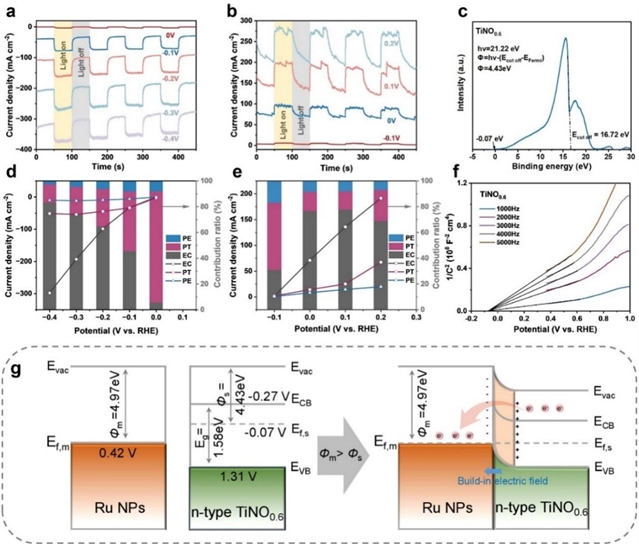
图5. 光热(PT)与光电子(PE)效应的量化。(a-b) 在碱性海水溶液中,不同电位下近红外光照射Ru/TiNO0.6电极产生的HER和HzOR光电流响应。(c) TiNO0.6的UPS谱图。(d, e) 在不同施加电位的光照条件下,PE、PT和EC(电化学)的电流密度及其对HER和HzOR的贡献百分比。(f) TiNO0.6的Mott-Schottky曲线。(g) RuNPs和TiNO0.6的能带图。
通过光电性能测试系统分析了等离激元增强机制。光电流响应测试(图5a-b)显示,在低过电位下光热效应贡献显著(HER达86.3%,HzOR达52.1%),但随着过电位增加,电化学贡献逐渐占据主导。UPS谱图(图5c)确定TiNO0.6的功函数为4.43 eV。Mott-Schottky曲线(图5f)显示n型半导体特性,平带电位为-0.14 V。能带结构示意图(图5g)表明,Ru与TiNO0.6形成Mott-Schottky异质结,产生从TiNO0.6指向Ru的内建电场,促进界面电荷分离。
6.中间体与反应机理分析
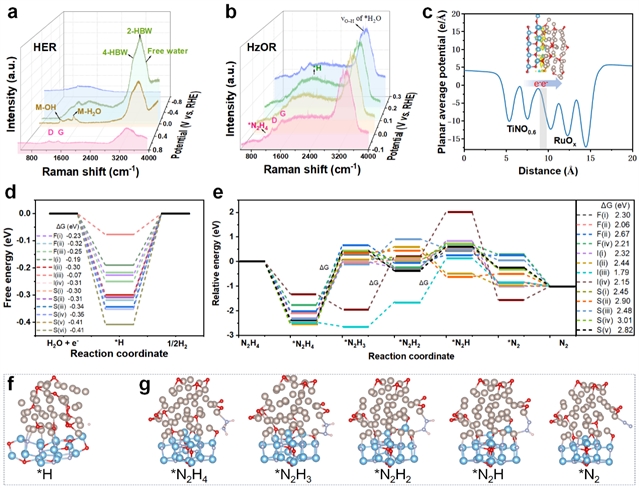
图6. 中间体与反应机理分析。(a, b) 在HER和HzOR过程中,Ru/TiNO0.6电极上原位拉曼光谱。(c) Ru/TiNO0.6异质结模型的平面平均电势和电荷密度差(CDD)图,其中青色和黄色区域分别表示电荷的耗尽和积累。(d, e) 在不同活性位点上,HER的H吸附和HzOR的NxH的自由能变化计算图。(f, g) 分别在I(iii)模型上HER的H中间体和HzOR的NxHy中间体吸附示意图。
原位拉曼光谱揭示了反应过程中的中间体演化(图6a-b)。在HER过程中,随着电位负移,M-OH(1126 cm-1)和M-H2O(1515 cm-1)信号增强,表明水解离加速。在HzOR过程中,N2H4特征峰(1127 cm-1)在开路电位下即出现,证实催化剂的本征肼分解活性。DFT计算显示(图6c-g),界面处的电荷重新分布优化了*H吸附能(ΔGH ≈ -0.07 eV),并将HzOR的速率决定步骤从N2H4→*N2H3转变为*N2H2→*N2H,显著降低反应能垒。
五、启示
本研究采用氮气作为“分子开关”来激发Ru/TiNO0.6莫特-肖特基异质结中的等离激元双功能特性。在近红外光照射下,HER和HzOR的过电位分别降低至约13.5 mV和222.3 mV,使得等离激元增强的双电极电解在0.2 V下实现电流密度从127.5 mA cm-2提升至171.6 mA cm-2的OHzS,同时具有对H2/N2的高选择性和改进的稳定性。机理分析揭示了氮气的三重作用:缩小基底带隙并增强光电效应和光热效应;增强莫特-肖特基效应以生成亚稳态非晶态Ru,同时诱导界面电荷极化并产生协同降低活化能垒的内建电场。这些协同效应在促进N2H3中间体形成和RDS位移的同时,实现了最优的氢吸附能(ΔG*H)。本研究通过等离激元耦合肼-海水电解展示了先进的氢气产出路线,为新型能源转换技术的开发和应用铺平了道路。
引用信息:Jiajun Luo, Congyuan Zeng, Rui Yang, Jingyun Mao, Hui Xue, Shuangjuan Shen, Yiyin Huang, Yuanyuan Sun, Haoran Jiang, Yaobing Wang, Nitrogen-triggered plasmonic bifunctionality in ruthenium/titanium oxynitride Schottky catalyst for energized hydrazine-seawater hydrogen production, Adv. Powder Mater. 5(2026)100384. https://doi.org/10.1016/j.apmate.2025.100384

扫二维码 查看全文
原文链接:https://www.sciencedirect.com/science/article/pii/S2772834X25001204
特别声明:本文转载仅仅是出于传播信息的需要,并不意味着代表本网站观点或证实其内容的真实性;如其他媒体、网站或个人从本网站转载使用,须保留本网站注明的“来源”,并自负版权等法律责任;作者如果不希望被转载或者联系转载稿费等事宜,请与我们接洽。






