|
|
|
|
|
3D Genome Browserпјҡе®һзҺ°еҸҜи§ҶеҢ–еҹәеӣ з»„3Dз»“жһ„е’Ңй•ҝи·қзҰ»жҹ“иүІдҪ“дҪңз”Ё | Genome Biology |
|
|
и®әж–Үж ҮйўҳпјҡThe 3D Genome Browser: a web-based browser for visualizing 3D genome organization and long-range chromatin interactions
жңҹеҲҠпјҡ Genome Biology
дҪңиҖ…пјҡ Yanli Wang, Fan Song, Bo Zhang, Lijun Zhang, Jie Xu, Da Kuang, Daofeng Li, Mayank N. K. Choudhary, Yun Li, Ming Hu, Ross Hardison, Ting Wang and Feng Yue
еҸ‘иЎЁж—¶й—ҙпјҡ2018/10/4
ж•°еӯ—иҜҶеҲ«з Ғпјҡ 10.1186/s13059-018-1519-9
еҺҹж–Үй“ҫжҺҘпјҡhttps://genomebiology.biomedcentral.com/articles/10.1186/s13059-018-1519-9?utm_source=WeChat&utm_medium=Website_linksSocial_media_organic&utm_content=CelZha-MixedBrand-multijournal-Multidisciplinary-China&utm_campaign=ORG_AWA_CZH_BMCWechat_dailyposts_blogs
еҫ®дҝЎй“ҫжҺҘпјҡhttps://mp.weixin.qq.com/s/oTjxIH9cPhrgrCBlOOLRaw
еңЁе“әд№іеҠЁзү©зҡ„еҹәеӣ з»„дёӯпјҢжҹ“иүІдҪ“зҡ„дёүз»ҙз»“жһ„еңЁеҹәеӣ иЎЁиҫҫи°ғжҺ§дёӯжү®жј”зқҖйҮҚиҰҒзҡ„и§’иүІгҖӮеңЁDNAеұӮйқўдёҠпјҢиҝңз«Ҝзҡ„и°ғжҺ§еҺҹ件пјҲеҰӮеўһејәеӯҗпјүеҸҜд»ҘйҖҡиҝҮз©әй—ҙз»“жһ„зҡ„жҺҘиҝ‘жқҘи°ғжҺ§зӣ®ж Үеҹәеӣ зҡ„иЎЁиҫҫгҖӮеңЁжӣҙй«ҳдёҖзә§зҡ„жҹ“иүІдҪ“з»“жһ„дёӯпјҢиҝ‘е№ҙжқҘеҸ‘зҺ°зҡ„жӢ“жү‘з»“жһ„еҹҹпјҲtopologically associating domainsпјүиў«и®ӨдёәжҳҜе“әд№іеҠЁзү©жҹ“иүІдҪ“з»“жһ„зҡ„еҹәжң¬еҚ•е…ғ[1]гҖӮжӢ“жү‘з»“жһ„еҹҹжҳҜеӨ§е°Ҹдёәе…Ҷзўұеҹәзҡ„жҹ“иүІдҪ“й«ҳзә§з»“жһ„д№ӢдёҖпјҢжңҖж—©иў«Hi-CжҠҖжңҜжүҖжҸӯзӨәгҖӮжӢ“жү‘з»“жһ„еҹҹдёәи§ЈйҮҠиҝңз«Ҝеўһејәеӯҗзҡ„еҹәеӣ и°ғжҺ§жҸҗдҫӣдәҶйқһеёёеҘҪзҡ„и§ЈйҮҠгҖӮйҷӨдәҶжӢ“жү‘з»“жһ„еҹҹпјҢе…¶д»–жҹ“иүІдҪ“з»“жһ„дҫӢеҰӮA/Bз»“жһ„[2]гҖҒжҹ“иүІдҪ“зҺҜпјҲchromatin loopпјү[3]зӯүд№ҹзӣёеә”иў«Hi-CиЎҚз”ҹжҠҖжңҜжүҖжҸӯзӨәгҖӮиҝҷдәӣHi-CиЎҚз”ҹжҠҖжңҜпјҢдҫӢеҰӮHi-CпјҢChIA-PET[4]пјҢCapture Hi-C[5]пјҢPLAC-seq[6]е’ҢHiChIP[7]пјҢдёәз ”з©¶жҹ“иүІдҪ“з©әй—ҙз»“жһ„жҸҗдҫӣдәҶеүҚжүҖжңӘжңүзҡ„жңәдјҡе’ҢжҢ‘жҲҳгҖӮ然иҖҢпјҢйҡҸзқҖHi-Cж•°жҚ®зҡ„йЈһйҖҹеўһй•ҝпјҢй«ҳж•ҲдҫҝеҲ©зҡ„ж•°жҚ®еҸҜи§ҶеҢ–жҠҖжңҜеҸҳеҫ—иҝ«еңЁзңүзқ«гҖӮеҗҢж—¶пјҢй«ҳж•Ҳзҡ„еҸҜи§ҶеҢ–еҜ№дәҺжҸӯзӨәйҡҗи—ҸеңЁHi-Cж•°жҚ®иғҢеҗҺзҡ„з”ҹзү©еӯҰж„Ҹд№үе…·жңүйҮҚиҰҒдҪңз”ЁгҖӮ然иҖҢпјҢз”ұдәҺиҝҷдәӣж•°жҚ®йҖҡеёёж•°йҮҸеәһеӨ§дё”еӨҚжқӮпјҢзӢ¬з«Ӣз ”з©¶иҖ…еҰӮжһңжғіиҮӘе·ұе®һзҺ°ж•°жҚ®зҡ„еҸҜи§ҶеҢ–е°ҶеҸҳеҫ—дҪҺж•Ҳдё”иҖ—ж—¶гҖӮ
жңҖиҝ‘жқҘиҮӘзҫҺеӣҪе®ҫе·һе·һз«ӢеӨ§еӯҰзҡ„еІіеі°иҜҫйўҳз»„е’ҢеҚҺзӣӣйЎҝеӨ§еӯҰзҡ„зҺӢиүҮиҜҫйўҳз»„еңЁGenome BiologyеҸ‘иЎЁдәҶдёҖзҜҮеҗҚдёә“The 3D Genome Browser: a web-based browser for visualizing 3D genome organization and long-range chromatin interactions”зҡ„ж–№жі•ж–Үз« гҖӮиҜҘж–Үз« д»Ӣз»ҚдәҶ他们ејҖеҸ‘зҡ„еҹәдәҺзҪ‘йЎөзҡ„3Dеҹәеӣ з»„жөҸи§ҲеҷЁпјҲwww.3dgenome.orgпјүгҖӮиҝҷжҳҜзӣ®еүҚжңҖжөҒиЎҢзҡ„3Dеҹәеӣ з»„жөҸи§ҲеҷЁпјҢиҝ„д»ҠдёәжӯўпјҢ е·Іиў«жқҘиҮӘ120еӨҡдёӘеӣҪ家зҡ„ж•°дёҮи®Ўз”ЁжҲ·и®ҝй—®иҝҮпјҢзҪ‘йЎөзӮ№еҮ»йҮҸе·Із»Ҹи¶…иҝҮдәҶж•°еҚҒдёҮж¬ЎгҖӮиҜҘзҪ‘з«ҷжӢҘжңүдј—еӨҡжҹ“иүІдҪ“з»“жһ„зӣёе…іж•°жҚ®зұ»еһӢпјҢеҢ…жӢ¬Hi-CпјҢGAMпјҢSPRITEпјҢDNase Hi-CпјҢChIA-PETпјҢPLAC-seqпјҢHiChIPе’ҢCapture Hi-CгҖӮж•°жҚ®еҜ№иұЎеҢ…жӢ¬дәәзұ»е’Ңе°Ҹйј зҡ„ж•°еҚҒз§Қз»„з»Үе’Ңз»Ҷиғһзі»пјҢжҖ»е…ұж•°жҚ®йҮҸиҫҫеҲ°300дҪҷдёӘгҖӮзҪ‘з«ҷж•°жҚ®еҠ иҪҪиҝ…йҖҹпјҢеҸҜд»ҘеңЁ5з§’д№ӢеҶ…жү“ејҖдёҖдёӘ10MbеӨ§е°ҸеҢәеҹҹзҡ„Hi-CзғӯеӣҫгҖӮ
иҜҘжөҸи§ҲеҷЁиғҪеӨҹдҪҝз ”з©¶дәәе‘ҳжӣҙеҠ ж–№дҫҝеҝ«жҚ·ең°еҸҜи§ҶеҢ–жқҘиҮӘдәҺй«ҳйҖҡйҮҸжҹ“иүІдҪ“з»“жһ„жҚ•иҺ·жҠҖжңҜпјҲHi-Cпјүзҡ„ж•°жҚ®гҖӮе…¶дё»иҰҒеҠҹиғҪеҰӮдёӢпјҡ1пјүдҪҝз”Ёзғӯеӣҫзҡ„ж–№ејҸеҸҜи§ҶеҢ–Hi-Cж•°жҚ®д»ҘеҸҠHi-Cзұ»еһӢжҠҖжңҜеҰӮGAMгҖҒSPRITEгҖҒе’ҢDNase Hi-Cпјӣ2пјүеҸҜи§ҶеҢ–дёҚеҗҢжҹ“иүІдҪ“д№Ӣй—ҙзҡ„3Dз»“жһ„пјӣ3пјүжҜ”иҫғдёҚеҗҢз»„з»ҮжҲ–иҖ…зү©з§Қд№Ӣй—ҙзҡ„жҹ“иүІдҪ“3Dжһ„еһӢпјӣ4пјүе°ҶHi-C ж•°жҚ®иҪ¬жҚўдёәиҷҡжӢҹ4CпјҲvirtual 4CпјүпјҢд»ҺиҖҢеҸҜд»Ҙжӣҙж–№дҫҝең°жҹҘзңӢзү№е®ҡдҪҚзӮ№е’Ңжҹ“иүІдҪ“дёҠе…¶д»–дҪҚзҪ®зҡ„зӣёдә’дҪңз”Ёпјӣ5пјүеҸҜи§ҶеҢ–еҹәдәҺжҹ“иүІиҙЁе…Қз–«е…ұжІүж·Җз»“еҗҲжҲ–жҹ“иүІдҪ“еҢәеҹҹжҚ•иҺ·зҡ„Hi-CиЎҚз”ҹжҠҖжңҜпјҲдҫӢеҰӮChIA-PETпјҢPLAC-seqпјҢ HiChIPпјҢCapture Hi-CпјүгҖӮ
еңЁиҝҷдёӘйЎ№зӣ®йҮҢпјҢз ”з©¶е‘ҳ们жҸҗеҮәдәҶдёҖз§Қж–°зҡ„дәҢиҝӣеҲ¶ж јејҸпјҲBUTLRпјүз”ЁжқҘдҝқеӯҳHi-Cж•°жҚ®пјҢд»ҺиҖҢжһҒеӨ§зҡ„еҮҸе°‘дҪҝж–Ү件еӨ§е°Ҹ并且жҸҗй«ҳжҹҘиҜўйҖҹеәҰгҖӮйҷӨжӯӨд№ӢеӨ–пјҢиҜҘBrowserиҝҳжҸҗдҫӣдәҶж–№дҫҝзҡ„жҹ“иүІдҪ“еҢәеҹҹзј©ж”ҫеҠҹиғҪгҖӮз”ЁжҲ·еҸҜд»ҘйҖҡиҝҮеҹәеӣ пјҢжҹ“иүІдҪ“дҪҚзҪ®пјҢе’ҢSNPзј–еҸ·еҜ№Hi-Cж•°жҚ®иҝӣиЎҢжҹҘиҜўгҖӮжӣҙдёәйҮҚиҰҒзҡ„жҳҜпјҢз”ЁжҲ·еҸҜд»Ҙе°ҶиҮӘе·ұзҡ„UCSCжҲ–иҖ…WashU trackе’ҢHi-CзғӯеӣҫиҝӣиЎҢж— зјқиЎ”жҺҘпјҢд»ҺиҖҢжһҒеӨ§ең°жү©еұ•дәҶеҸҜд»ҘжҳҫзӨәең°ж•°жҚ®зұ»еһӢгҖӮ
дёӢйқўжҲ‘们е°Ҷз”ЁеҮ дёӘдҫӢеӯҗжқҘиҜҙжҳҺеҰӮдҪ•дҪҝз”Ё3D Genome BrowserиҝӣиЎҢHi-Cж•°жҚ®жҢ–жҺҳгҖӮ

еӣҫ1: 3D еҹәеӣ з»„жөҸи§ҲеҷЁзҡ„Hi-CжҹҘиҜўйЎөйқўгҖӮз”ЁжҲ·еҸҜд»ҘйҖүжӢ©зҪ‘з«ҷеҶ…е»әж•°жҚ®пјҢжҲ–иҖ…дёҠдј иҮӘе·ұеҲ¶дҪңзҡ„BUTLRж–Ү件гҖӮ
1. еҲ©з”ЁHi-Cж•°жҚ®з ”究жҹ“иүІдҪ“зӣёдә’дҪңз”Ё
йҰ–е…ҲпјҢжү“ејҖHi-CжҹҘиҜўйЎөйқўпјҲеӣҫ1пјүпјҢеңЁиҜҘйЎөйқўдёӯжҲ‘们еҸҜд»ҘйҖүжӢ©Hi-Cж–№жі•гҖҒзү©з§ҚгҖҒеҸӮз…§еҹәеӣ з»„гҖҒз»„з»ҮжҲ–з»Ҷиғһзі»д»ҘеҸҠж•°жҚ®еҲҶиҫЁзҺҮгҖӮдҫӢеҰӮпјҢжҲ‘们еҸҜд»ҘжҹҘиҜўSHHеҹәеӣ еңЁGM12878з»Ҷиғһзі»дёӯзҡ„Hi-CзғӯеӣҫгҖӮд»ҺHi-CзғӯеӣҫдёҠпјҲеӣҫ2пјүжҲ‘们еҸҜд»ҘзңӢеҮәпјҢиҜҘеҹәеӣ дёҺе…¶дёҠжёёдёҖдёӘе·ІзҹҘзҡ„еўһејәеӯҗеҢәеҹҹдҪҚдәҺеҗҢдёҖTADеҶ…пјҢ并且иҜҘеўһејәеӯҗе’ҢSHHеҗҜеҠЁеӯҗеӯҳеңЁиҫғй«ҳзҡ„зӣёдә’дҪңз”Ё,д»ҺиҖҢиҜҒе®һдәҶиҜҘеўһејәеӯҗеҜ№SHHзҡ„и°ғжҺ§дҪңз”ЁгҖӮжҲ‘们иҝҳеҸҜд»ҘзӮ№еҮ»йЎөйқўеҸідёҠж–№зҡ„жҹұзҠ¶еӣҫжҹҘзңӢиҜҘеҹәеӣ еңЁENCODEдёӯ100еӨҡдёӘз»„з»Үдёӯзҡ„иЎЁиҫҫжғ…еҶөгҖӮ

еӣҫ2: SHHеҹәеӣ еңЁGM12878з»Ҷиғһзі»дёӯзҡ„Hi-CзғӯеӣҫгҖӮиҷҡзәҝжҳҫзӨәSHHдёҺдёҠжёёдёҖдёӘеўһејәеӯҗеҢәеҹҹеӯҳеңЁй«ҳзӣёдә’дҪңз”ЁпјҢ并且иҜҘеўһејәеӯҗе’ҢSHHеңЁеҗҢдёҖдёӘTADдёӯгҖӮ
2. еҲ©з”ЁVirtual 4CгҖҒDHS linkageе’ҢChIA-PETз ”з©¶SNPзҡ„зӣ®ж Үеҹәеӣ
Hi-CиҝҳеҸҜд»ҘжҸӯзӨәSNPзҡ„жҪңеңЁзӣ®ж Үеҹәеӣ гҖӮжҲ‘们еҲ©з”Ёvirtual 4CжҹҘиҜўrs12740374пјҢиҜҘSNPдёҺдәәзҫӨдёӯдҪҺеҜҶеәҰи„ӮиӣӢзҷҪеҚҮй«ҳжңүе…ігҖӮиҷҡжӢҹ4CжҳҫзӨәиҜҘSNPдёҺе…¶дёӢжёёSORT1зҡ„еҗҜеҠЁеӯҗеӯҳеңЁиҫғй«ҳдҪңз”ЁпјҢиҝҷдёҖеҸ‘зҺ°еҗҢж—¶д№ҹиў«DNAй«ҳж•Ҹж„ҹжҖ§дҪҚзӮ№иҝһй”ҒпјҲDHS linkageпјүе’ҢChIA-PETж•°жҚ®жүҖж”ҜжҢҒпјҲеӣҫ3пјүгҖӮеҗҢж—¶еӣҫдёӯдёӢж–№зҡ„з»„иӣӢзҷҪж•°жҚ®д№ҹжҳҫзӨәиҜҘSNPдҪҚдәҺдёҖдёӘеҸҜиғҪзҡ„еўһејәеӯҗеҢәеҹҹгҖӮиҝҷдәӣиҜҒжҚ®иЎЁжҳҺSORT1еҸҜиғҪжҳҜrs12740374зҡ„зӣ®ж Үеҹәеӣ гҖӮ

еӣҫ3: еҲ©з”ЁиҷҡжӢҹ4Cе’ҢDHSиҝһй”ҒжҸӯзӨәSNPзҡ„жҪңеңЁзӣ®ж Үеҹәеӣ гҖӮrs12740374дҪҚдәҺдёҖдёӘеҸҜиғҪзҡ„еўһејәеӯҗеҢәеҹҹе’ҢCEBPBиӣӢзҷҪз»“еҗҲдҪҚзӮ№гҖӮ
3. еҲ©з”ЁCapture Hi-Cж•°жҚ®з ”究й«ҳеҲҶиҫЁзҺҮеҗҜеҠЁеӯҗ-еўһејәеӯҗзҡ„зӣёдә’дҪңз”Ё
еңЁеӣҫ4дёӯпјҢжҲ‘们еҲ©з”ЁCapture Hi-Cж•°жҚ®еҲҶжһҗPAX5зҡ„и°ғжҺ§жңәеҲ¶гҖӮжҲ‘们йҰ–е…ҲеҸ‘зҺ°еңЁеҺҹе§ӢBз»ҶиғһдёӯPAX5зҡ„еҗҜеҠЁеӯҗе’Ңе…¶дёҠжёёзҡ„ZCCHC7еҢәеҹҹеӯҳеңЁзӣёдә’дҪңз”Ё[13]гҖӮйҖҡиҝҮDNAй…¶и¶…ж•Ҹж„ҹдҪҚзӮ№пјҲDHSпјүж•°жҚ®е’Ңз»„иӣӢзҷҪдҝ®йҘ°ж•°жҚ®пјҢжҲ‘们д№ҹеҸ‘зҺ°дәҶдёҺиҜҘеҗҜеҠЁеӯҗдҪңз”Ёзҡ„еҢәеҹҹжҳҜдёҖдёӘеҸҜиғҪзҡ„еўһејәеӯҗгҖӮеҗҢж ·зҡ„пјҢеңЁHi-CзғӯеӣҫдёҠпјҢжҲ‘们д№ҹеҸ‘зҺ°дәҶиҝҷдёӨдёӘеҢәеҹҹеӯҳеңЁиҫғејәзҡ„зӣёдә’дҪңз”ЁгҖӮиҝҷдәӣиҜҒжҚ®йғҪиҜҙжҳҺдәҶдҪҚдәҺZCCHC7дёҠжёёзҡ„иҝҷдёӘеўһејәеӯҗеҸҜиғҪжҳҜи°ғжҺ§PAX5еҹәеӣ иЎЁиҫҫзҡ„е…ій”®гҖӮжңүи¶Јзҡ„жҳҜпјҢд№ӢеүҚзҡ„з ”з©¶жҠҘйҒ“жҢҮеҮәиҜҘеўһејәеӯҗзҡ„еҲ йҷӨдҪҝеҫ—PAX5иЎЁиҫҫдёӢи°ғ并еҜјиҮҙзҷҪиЎҖз—…[14]гҖӮиҝҷдёӘдҫӢеӯҗеҸҚжҳ дәҶжҲ‘们еҸҜд»ҘеҲ©з”Ё3D Genome Browserе’ҢдҪҝз”ЁCapture Hi-Cж•°жҚ®з ”究зІҫз»Ҷзҡ„еҗҜеҠЁеӯҗ-еўһејәеӯҗдҪңз”ЁгҖӮ

еӣҫ4: еҲ©з”ЁCapture Hi-Cж•°жҚ®жҸӯзӨәPAX5зҡ„еҸҜиғҪи°ғжҺ§жңәеҲ¶гҖӮ
4. йҖҡиҝҮеҜ№жҜ”Hi-CзғӯеӣҫжқҘз ”з©¶дёҚеҗҢзү©з§Қй—ҙжҹ“иүІиҙЁжһ„еһӢзҡ„дҝқе®ҲзЁӢеәҰ
жҲ‘们иҝҳеҸҜд»Ҙз”ЁHi-CеҜ№жҜ”жЁЎејҸжқҘз ”з©¶жҹ“иүІиҙЁжһ„еһӢеңЁдёҚеҗҢзү©з§Қд№Ӣй—ҙзҡ„дҝқе®ҲжҖ§гҖӮеӣҫ5жҳҫзӨәдәҶBCL6еҢәеҹҹеңЁдәәж·Ӣе·ҙз»Ҷиғһзі»пјҲGM12878пјүе’Ңйј з»Ҷиғһзі»пјҲCH12пјүзҡ„Hi-CзғӯеӣҫгҖӮд»ҺHi-Cзғӯеӣҫзҡ„зӣёдјјжҖ§еҸҜд»ҘзңӢеҮәиҜҘеҢәеҹҹзҡ„жҹ“иүІиҙЁжһ„еһӢеңЁдәәе’Ңйј дёӯдҝқе®ҲзЁӢеәҰиҫғй«ҳгҖӮ

еӣҫ5: жҜ”иҫғдәәе’Ңе°Ҹйј зҡ„BCL6еҹәеӣ йҷ„иҝ‘зҡ„жҹ“иүІиҙЁжһ„еһӢзҡ„дҝқе®ҲжҖ§гҖӮ
5. еҲ©з”ЁHi-Cж•°жҚ®жЈҖжөӢжҹ“иүІдҪ“з»“жһ„еҸҳејӮ
и®ёеӨҡз ”з©¶еҸ‘зҺ°Hi-CеҸҜд»Ҙиў«з”ЁдәҺжЈҖжөӢжҹ“иүІдҪ“з»“жһ„еҸҳејӮпјҲstructural variantionпјүгҖӮдёҚеҗҢзұ»еһӢзҡ„з»“жһ„еҸҳејӮпјҢдҫӢеҰӮеҲ еӨұгҖҒжҸ’е…ҘгҖҒејӮдҪҚе’ҢеҖ’иЈ…йғҪдјҡеҜјиҮҙHi-CзғӯеӣҫеҸ‘з”ҹзӣёеә”зҡ„еҸҳеҢ–[15]гҖӮBCR-ABLеҹәеӣ иһҚеҗҲжҳҜж…ўжҖ§зІ’зі»зҷҪиЎҖз—…зҡ„иҮҙз—…еҸҳејӮгҖӮиҜҘеҸҳејӮжҳҜз”ұ9еҸ·жҹ“иүІдҪ“е’Ң22еҸ·жҹ“иүІдҪ“зҡ„ејӮдҪҚеҸҳејӮеҜјиҮҙзҡ„гҖӮжҲ‘们еҸҜд»ҘйҖҡиҝҮHi-CзғӯеӣҫжЈҖжөӢиҜҘејӮдҪҚеҸҳејӮпјҲеӣҫ6пјүгҖӮK562жҳҜд»Һж…ўжҖ§зІ’зі»зҷҪиЎҖз—…жӮЈиҖ…дёӯеҹ№иӮІзҡ„з»Ҷиғһзі»гҖӮеӣҫ6aжҳҫзӨәзҡ„жҳҜK562дёӯ22еҸ·жҹ“иүІдҪ“9еҸ·жҹ“иүІдҪ“зҡ„Hi-CзғӯеӣҫгҖӮдёӯй—ҙиҸұеҪўеҢәеҹҹжҳҫзӨәеҮә22еҸ·жҹ“иүІдҪ“9еҸ·жҹ“иүІдҪ“еӯҳеңЁиҫғејәзҡ„зӣёдә’е·ҰеҸіпјҢиҖҢеңЁжӯЈеёёз»ҶиғһдёӯпјҢиҝҷз§Қжҹ“иүІдҪ“д№Ӣй—ҙзҡ„зӣёдә’дҪңз”ЁжҳҜеҫ®д№Һе…¶еҫ®зҡ„пјҢ并且иҝңе°ҸдәҺжҹ“иүІдҪ“еҶ…йғЁзҡ„зӣёдә’дҪңз”ЁгҖӮеӣ жӯӨпјҢжҲ‘们иғҪеӨҹжҺЁж–ӯеҮә22еҸ·жҹ“иүІдҪ“9еҸ·жҹ“иүІдҪ“еҸ‘з”ҹдәҶиһҚеҗҲпјҢеҜјиҮҙдәҶдёҚеҗҢжҹ“иүІдҪ“д№Ӣй—ҙзҡ„зӣёдә’дҪңз”ЁеўһејәгҖӮ并且еңЁиһҚеҗҲж–ӯзӮ№еҢәеҹҹпјҢжҲ‘们еҸҜд»ҘзңӢеҲ°жҳҜBCRе’ҢABLеҹәеӣ еҸ‘з”ҹдәҶиһҚеҗҲгҖӮзӣёжҜ”д№ӢдёӢпјҢеңЁжӯЈеёёзҡ„GM12878ж·Ӣе·ҙз»Ҷиғһзі»дёӯпјҢжҲ‘们没жңүзңӢеҲ°зұ»дјјзҡ„дёҚеҗҢжҹ“иүІдҪ“д№Ӣй—ҙзҡ„зӣёдә’дҪңз”ЁеўһејәпјҲеӣҫ6b)гҖӮйҖҡиҝҮ3D Genome Browserзҡ„жҹҘиҜўжҹ“иүІдҪ“д№Ӣй—ҙзҡ„дҪңз”ЁеҠҹиғҪпјҢз ”з©¶иҖ…иғҪеӨҹеҸ‘зҺ°еҹәеӣ з»„зҡ„з»“жһ„еҸҳејӮеҸҠе…¶еҸҜиғҪеҜјиҮҙзҡ„еҹәеӣ и°ғжҺ§ж”№еҸҳгҖӮ
3D Genome Browserиҝҳжңүдј—еӨҡеҠҹиғҪжңӘиғҪеңЁжң¬ж–Үж¶өзӣ–пјҢдҫӢеҰӮиҷҡжӢҹ4CпјҲvirtual 4Cпјүе’ҢдёҚеҗҢHi-Cж•°жҚ®зҡ„е·®ејӮеҲҶжһҗпјҢиҝҷдәӣеҠҹиғҪеҗҢж ·жңүеҠ©дәҺжҢ–жҺҳHi-Cж•°жҚ®иғҢеҗҺзҡ„з”ҹзү©еӯҰж„Ҹд№үгҖӮж¬ўиҝҺе№ҝеӨ§жңүеҝ—дәҺеӯҰд№ 3Dеҹәеӣ з»„зҡ„з§‘з ”е·ҘдҪңиҖ…дҪҝз”Ёе’ҢжҺўзҙўжҲ‘们зҡ„3D Genome BrowserгҖӮ
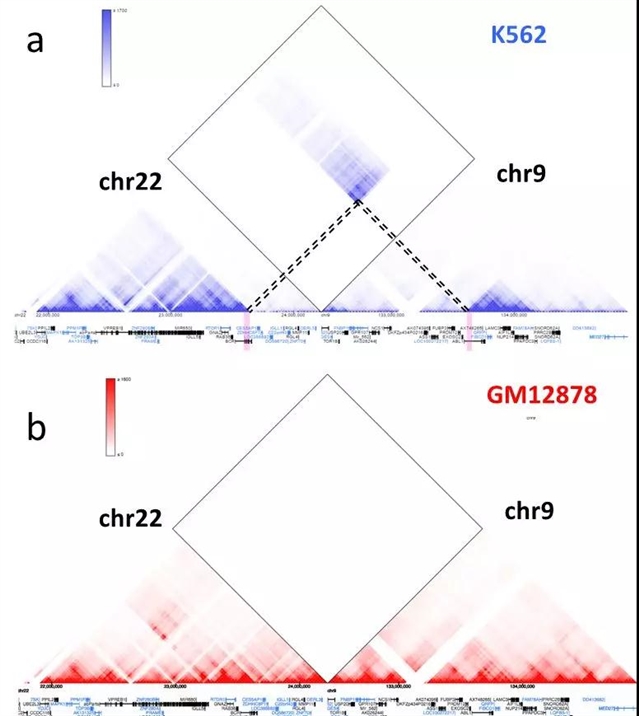
еӣҫ6: a).K562дёӯchr22е’Ңchr9зҡ„Hi-CзғӯеӣҫпјҢиҷҡзәҝжҳҫзӨәж–ӯзӮ№еңЁдёӨжқЎжҹ“иүІдҪ“жүҖеңЁдҪҚзҪ®гҖӮb).GM12878дёӯжӯЈеёёзҡ„chr22е’Ңchr9зҡ„Hi-CзғӯеӣҫжІЎжңүжҳҫзӨәеҮәеўһејәзҡ„жҹ“иүІдҪ“д№Ӣй—ҙзҡ„зӣёдә’дҪңз”ЁгҖӮ
еҸӮиҖғж–ҮзҢ®пјҡ
гҖҗ1гҖ‘ Dixon JR, Selvaraj S, Yue F, Kim A, Li Y, Shen Y, Hu M, Liu JS, Ren B: Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature 2012, 485:376-380.
гҖҗ2гҖ‘ Lieberman-Aiden E, van Berkum NL, Williams L, Imakaev M, Ragoczy T, Telling A, Amit I, Lajoie BR, Sabo PJ, Dorschner MO, et al: Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 2009, 326:289-293.
гҖҗ3гҖ‘ Rao SS, Huntley MH, Durand NC, Stamenova EK, Bochkov ID, Robinson JT, Sanborn AL, Machol I, Omer AD, Lander ES, Aiden EL: A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 2014, 159:1665-1680.
гҖҗ4гҖ‘ Li G, Fullwood MJ, Xu H, Mulawadi FH, Velkov S, Vega V, Ariyaratne PN, Mohamed YB, Ooi HS, Tennakoon C, et al: ChIA-PET tool for comprehensive chromatin interaction analysis with paired-end tag sequencing. Genome Biol 2010, 11:R22.
гҖҗ5гҖ‘ Mifsud B, Tavares-Cadete F, Young AN, Sugar R, Schoenfelder S, Ferreira L, Wingett SW, Andrews S, Grey W, Ewels PA, et al: Mapping long-range promoter contacts in human cells with high-resolution capture Hi-C. Nat Genet 2015, 47:598-606.
гҖҗ6гҖ‘ Fang R, Yu M, Li G, Chee S, Liu T, Schmitt AD, Ren B: Mapping of long-range chromatin interactions by proximity ligation-assisted ChIP-seq. Cell Res 2016, 26:1345-1348.
гҖҗ7гҖ‘ Mumbach MR, Rubin AJ, Flynn RA, Dai C, Khavari PA, Greenleaf WJ, Chang HY: HiChIP: efficient and sensitive analysis of protein-directed genome architecture. Nat Methods 2016, 13:919-922.
гҖҗ8гҖ‘ Zhou X, Maricque B, Xie M, Li D, Sundaram V, Martin EA, Koebbe BC, Nielsen C, Hirst M, Farnham P, et al: The Human Epigenome Browser at Washington University. Nat Methods 2011, 8:989-990.
гҖҗ9гҖ‘ Durand NC, Robinson JT, Shamim MS, Machol I, Mesirov JP, Lander ES, Aiden EL: Juicebox Provides a Visualization System for Hi-C Contact Maps with Unlimited Zoom. Cell Syst 2016, 3:99-101.
гҖҗ10гҖ‘ Kerpedjiev P, Abdennur N, Lekschas F, McCallum C, Dinkla K, Strobelt H, Luber JM, Ouellette SB, Ahzir A, Kumar N, et al: HiGlass: Web-based Visual Comparison And Exploration Of Genome Interaction Maps. bioRxiv 2017.
гҖҗ11гҖ‘ Tang B, Li F, Li J, Zhao W, Zhang Z: Delta: a new web-based 3D genome visualization and analysis platform. Bioinformatics 2018, 34:1409-1410.
гҖҗ12гҖ‘ Huang J, Liu X, Li D, Shao Z, Cao H, Zhang Y, Trompouki E, Bowman TV, Zon LI, Yuan GC, et al: Dynamic Control of Enhancer Repertoires Drives Lineage and Stage-Specific Transcription during Hematopoiesis. Dev Cell 2016, 36:9-23.
гҖҗ13гҖ‘ Javierre BM, Burren OS, Wilder SP, Kreuzhuber R, Hill SM, Sewitz S, Cairns J, Wingett SW, Varnai C, Thiecke MJ, et al: Lineage-Specific Genome Architecture Links Enhancers and Non-coding Disease Variants to Target Gene Promoters. Cell 2016, 167:1369-1384 e1319.
гҖҗ14гҖ‘ Puente XS, Bea S, Valdes-Mas R, Villamor N, Gutierrez-Abril J, Martin-Subero JI, Munar M, Rubio-Perez C, Jares P, Aymerich M, et al: Non-coding recurrent mutations in chronic lymphocytic leukaemia.Nature 2015, 526:519-524.
гҖҗ15гҖ‘ Dixon J, Xu J, Dileep V, Zhan Y, Song F, Le VT, Yardimci GG, Chakraborty A, Bann DV, Wang Y, et al: An Integrative Framework For Detecting Structural Variations In Cancer Genomes. Nature Genetics2018.
ж‘ҳиҰҒпјҡ
Abstract
Here, we introduce the 3D Genome Browser, http://3dgenome.org, which allows users to conveniently explore both their own and over 300 publicly available chromatin interaction data of different types. We design a new binary data format for Hi-C data that reduces the file size by at least a magnitude and allows users to visualize chromatin interactions over millions of base pairs within seconds. Our browser provides multiple methods linking distal cis-regulatory elements with their potential target genes. Users can seamlessly integrate thousands of other omics data to gain a comprehensive view of both regulatory landscape and 3D genome structure.
йҳ…иҜ»и®әж–Үе…Ёж–ҮиҜ·и®ҝй—®пјҡhttps://genomebiology.biomedcentral.com/articles/10.1186/s13059-018-1519-9?utm_source=WeChat&utm_medium=Website_linksSocial_media_organic&utm_content=CelZha-MixedBrand-multijournal-Multidisciplinary-China&utm_campaign=ORG_AWA_CZH_BMCWechat_dailyposts_blogs
жңҹеҲҠд»Ӣз»Қпјҡ
Genome Biology(https://genomebiology.biomedcentral.com/) publishes outstanding research in all areas of biology and biomedicine studied from a genomic and post-genomic perspective.
The current impact factor is 13.214* and the journal is ranked 4th among research journals in the Genetics and Heredity category by Thomson Reuters. Genome Biology is the highest ranked open access journal in the category.
2017 Journal Metrics
Citation Impact
13.2 - 2-year Impact Factor
16.5 - 5-year Impact Factor
3.1 - Source Normalized Impact per Paper (SNIP)
12.7 - SCImago Journal Rank (SJR)
пјҲжқҘжәҗпјҡ科еӯҰзҪ‘пјү
зү№еҲ«еЈ°жҳҺпјҡжң¬ж–ҮиҪ¬иҪҪд»…д»…жҳҜеҮәдәҺдј ж’ӯдҝЎжҒҜзҡ„йңҖиҰҒпјҢ并дёҚж„Ҹе‘ізқҖд»ЈиЎЁжң¬зҪ‘з«ҷи§ӮзӮ№жҲ–иҜҒе®һе…¶еҶ…е®№зҡ„зңҹе®һжҖ§пјӣеҰӮе…¶д»–еӘ’дҪ“гҖҒзҪ‘з«ҷжҲ–дёӘдәәд»Һжң¬зҪ‘з«ҷиҪ¬иҪҪдҪҝз”ЁпјҢйЎ»дҝқз•ҷжң¬зҪ‘з«ҷжіЁжҳҺзҡ„“жқҘжәҗ”пјҢ并иҮӘиҙҹзүҲжқғзӯүжі•еҫӢиҙЈд»»пјӣдҪңиҖ…еҰӮжһңдёҚеёҢжңӣиў«иҪ¬иҪҪжҲ–иҖ…иҒ”зі»иҪ¬иҪҪзЁҝиҙ№зӯүдәӢе®ңпјҢиҜ·дёҺжҲ‘们жҺҘжҙҪгҖӮ






