
近日,复旦大学徐昕团队研究了分子和晶体张量性质几何深度学习的通用框架。这一研究成果发表在2025年12月9日出版的《美国化学会志》上。
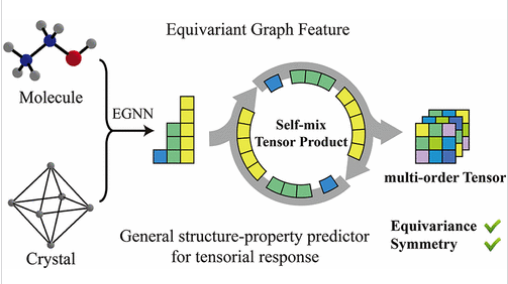
分子与晶体的响应特性天然地由遵循特定等变性和对称性约束的张量所描述。然而,机器学习模型直接预测这些张量仍然面临挑战。
研究组提出了一种用于等变图神经网络的通用输出模块,该模块能够以端到端的方式预测具有指定置换(基本)对称性的任意阶张量。结合SE(3)等变的XPaiNN架构,该框架实现了媲美第一性原理计算的精度,并且支持在一个统一的模型中预测原子尺度性质,如化学屏蔽张量和玻恩有效电荷。此外,该方法能够处理高阶张量,包括分子超极化率和晶体材料的弹性张量(刚度矩阵),从而可以推导和分析丰富的各向异性信息,助力人工智能辅助的功能分子与材料发现与设计。
附:英文原文
Title: General Framework for Geometric Deep Learning on Tensorial Properties of Molecules and Crystals
Author: Wenjie Yan, Xinming Lai, Yicheng Chen, Wenhao Zhang, Jianming Wu, Xin Xu
Issue&Volume: December 9, 2025
Abstract: Response properties of molecules and crystals are naturally described by tensors that obey specific equivariance and symmetry constraints. However, directly predicting these tensorial quantities remains challenging for machine learning models. We present a general-purpose output module for equivariant graph neural networks that enables end-to-end prediction of tensors of arbitrary order with prescribed permutation (fundamental) symmetry. Coupled with the SE(3)-equivariant XPaiNN architecture, our framework attains accuracy comparable to that of first-principles calculations. It also supports atomic-level properties─such as chemical shielding tensors and Born effective charges─in an all-in-one model. Moreover, the method handles higher-order tensors, including molecular hyperpolarizability and the elastic tensor (stiffness matrix) of crystalline materials, thereby enabling the derivation and analysis of rich anisotropic information and facilitating AI-assisted discovery and design of functional molecules and materials.
DOI: 10.1021/jacs.5c12428
Source: https://pubs.acs.org/doi/abs/10.1021/jacs.5c12428
JACS:《美国化学会志》,创刊于1879年。隶属于美国化学会,最新IF:16.383
官方网址:https://pubs.acs.org/journal/jacsat
投稿链接:https://acsparagonplus.acs.org/psweb/loginForm?code=1000