2022年11月9日,美国加州州立大学北岭分校孙远慧博士和苗茂生教授在Chem期刊上发表了一篇题为“Chemical templates that assemble the metal superhydrides”的研究成果。
该成果报道了一种全新的键合理论成功解释了金属超氢化合物的稳定性及其对不同金属的变化规律,并且发现大量结构中存在具有三维芳香性的氢单元,这为进一步发掘新型二元和多元金属超氢化合物结构提供了一个指导性方案。
论文第一作者是孙远慧;通讯作者是苗茂生。
金属氢化物超导是目前超导与高压领域的热点研究方向之一。在高压技术的帮助下,二元金属氢化物的理论和实验研究已几乎穷尽所有金属元素,发现了多种类型的金属超氢化合物结构。然而,普遍认为的金属与氢之间的化学键作用并非稳定结构中氢共价网络的化学驱动力。传统的化学键不能解释超氢化合物稳定性的变化规律。例如:为何元素周期表中s-d区边界处的金属更容易形成金属超氢化合物以及为何不同金属形成的超氢化合物会具有相同的结构原型?找到这一机理从而有效理解这些结构,对于不同金属超氢化物与其在不同压力下的稳定性和演化规律、以及进一步发掘新型二元及多元金属超氢化物材料具有重要意义。
近日,加州州立大学北岭分校孙远慧博士和苗茂生教授提出一种全新的键合理论成功解释了金属超氢化合物的稳定性及其对不同金属的变化规律。他们将许多报道过的金属超氢化合物结构拆分为金属子晶格和氢子晶格(共价网络)两部分,并通过电子结构计算发现这些金属子晶格的间隙位置处已经存在显著的电子占据。这些电子占据的位置刚好与放入的氢子晶格中氢原子(或氢单元)位置完全重合。金属轨道波函数分析表明金属子晶格的电子占据空隙处的定域轨道(准原子轨道),并在实空间中与氢晶格的未占据态具有类似的位置分布。因此当金属和氢的两个子晶格合并构成化合物时,金属子晶格占据准原子轨道的电子自然地占据氢晶格轨道,并被两者在一定程度上共享,从而降低体系的能量。作者们将这样的化学驱动力命名为“模板效应”。基于高通量第一性原理计算,作者证实元素周期表中s-d区边界处的金属所形成的化学模板效应最为显著,而目前报道的稳定金属超氢化合物中的金属也恰好来自该区域。自上世纪70年代以来,许多化学家都提出了以阴离子插入金属矩阵的角度理解化合物的思路,并以此解释了许多化合物的结构稳定性。本文的工作将这一半经验观点发展成基于准原子定域轨道的新型化学键理论,并以此成功解释了金属超氢化合物稳定性和结构规律。
基于此发现,作者利用具有较强“模板效应”的金属晶格作为初始结构,尝试设计新型超氢化合物结构,发现模板效应可以极大地提高搜索新型超氢化合物的效率。利用这一方法,他们发现并报道了一种Pbam SrH16二元相和一种P63/mmc Sc3MgH24三元相结构,理论计算预测的超导转变温度分别为138 K和40 K。
另外值得一提的是,氢晶格自身存在的芳香性氢单元也促进了结构的稳定。传统有机物结构中的芳香性官能团由碳骨架构成。其芳香性来自于碳原子垂直于C-C键方向的p轨道共轭形成的电子离域性。为保证需共轭的离域性,碳原子需要处在一个平面内并且满足4n+2规则。后者能够保证成键态被电子占满,而反键态全空的最低能量状态。和碳原子的p轨道不同,氢原子的1s电子成键不存在特定的方向性。相应的电子离域性也就无需被限制在平面结构中。相应地,3D氢单元最低能量态也不满足4n+2规则,而是决定于具体的结构。关键是实现成键态全部占据和反键态无占据的最低能量占据方式。作者们通过对不同氢单元的分析发现,大量具有芳香性的氢单元存在于超氢化合物结构中,其中包含以H8立方单元和H6冠状单元为代表的具有三维芳香性的氢单元。
文章的相关计算借助吉林大学材料科学与工程学院张立军教授团队开发的材料设计软件JAMIP和吉林大学物理学院马琰铭教授团队开发的晶体结构预测软件CALYPSO完成。本文第一作者孙远慧博士曾参与JAMIP的开发。

图1:金属超氢化合物的结构、稳定性和成键特性。(图片来源:Chem)
三种典型的金属超氢化合物结构:Im3m CaH6、Fm3m LaH10和P63/mmc CeH9。作者指出通常认为的电子转移、原子轨道成键等作用并非稳定金属超氢化合物结构的主要原因。

图2:金属晶格中电子在准原子轨道处的占据。(图片来源:Chem)
金属超氢化合物LaH10结构中的FCC La晶格的电子局域分布图,显示在其八面体心(EO)和四面体心(ET)位置处存在极值分布。更为深入的轨道波函数分析发现,这些极值的分布来自电子的占据态能级,即由金属晶格的准原子轨道所构成。这一点在许多稳定的金属超氢化合物结构中都存在。

图3:金属晶格中准原子占据的演化情况。(图片来源:Chem)
金属晶格的晶格参数在逐渐减小的过程中(外部压力上升),间隙处的电子会从低配位占据位置转移到高配位位置,对于不同金属,这一转移相应的晶格参数不同。通过对比元素周期表中同一周期的不同金属,发现早期金属的晶格间隙处的电子占据程度显著强于晚期金属。有趣的是,作者发现早期的Ca、Y和La等金属在形成超氢化合物结构时,对应的金属晶格在EO和ET位置处具有较大值;而对于晚期的金属,如Al,却并没有在这些位置处出现很强的电子占据。图3A和3B中阴影部分对应假象的Fm3m CaH10和Im3m CaH6结构在100-300 GPa压力下的晶格变化范围,图3E和3F中阴影部分对应假象的Fm3m AlH10和 Im3m AlH6结构结构在100-300 GPa压力下的晶格变化范围。

图4:超氢化合物结构中氢晶格的组成单元。(图片来源:Chem)
通过分析发现,氢晶格由多种不同类型的氢单元所构成。其中部分氢单元具有芳香性,例如图中的H6六边形、H8立方和H6冠状单元。由于这些芳香性氢单元的存在(图4D),导致氢晶格能量的显著降低。而其中以H8立方和H6冠状单元为代表的氢单元更是具有三维芳香性。与有机物结构中的碳原子π共轭条件下形成的芳香性不同,氢单元芳香性不需局限在平面内,其电子计数也不同于4n+2规则。
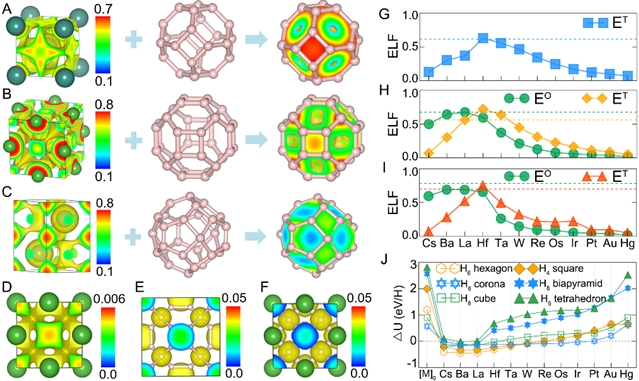
图5:金属模板协助氢晶格的形成。(图片来源:Chem)
金属超氢化合物中的金属晶格在间隙位置处具有电子占据,而当将氢晶格放回金属晶格中时,氢原子刚好坐落在电子占据分布的极值处。分析发现金属晶格的电子占据态能级与氢晶格的未占据态能级在实空间中具有相同的位置分布,因此二者在形成化合物时,金属晶格在氢晶格位置处的电子可以很顺利地转移给氢晶格,从而降低体系的能量。基于高通量第一性原理计算,作者证实元素周期表中s-d区边界处的金属所形成的化学驱动力最为明显,而目前报道的稳定金属超氢化合物结构中的金属也恰好来自该区域。作者将这样的化学驱动力命名为“模板效应”。

图6:“模板效应”对单金属超氢化合物结构稳定性的理解与预测。(图片来源:Chem)
“模板效应”理论对不同金属所形成的偏好超氢化合物结构具备很好的解释能力。当金属晶格的间隙位置处的准原子占据较强时,则偏向于形成这样的超氢化合物结构。基于此结论,作者利用具有较强“模板效应”的金属晶格作为起始结构,结合晶体结构预测方法,通过添加不同组分的氢原子来寻找可能存在的稳定金属超氢化合物结构。最终设计出一种Pbam SrH16结构,该结构在186 GPa以上开始稳定。此外,基于该理论,可快速并准确寻找到潜在的稳定结构,这些结构在常规的结构搜索设置下无法被探寻。

图7:“模板效应”对双金属超氢化合物的预测。(图片来源:Chem)
通过对已报道金属超氢化合物结构进行金属元素替换,可以构建出多种双金属超氢化合物结构。“模板效应”理论同样对其设计具有积极的指导能力。通过对比元素替换前后晶格中电子占据态的变化与相应形成能的变化,作者发现替换后电子占据程度增强的双金属超氢化合物结构的形成能大多是负值,这说明“模板效应”的增加有利于稳定双金属超氢化合物结构。同样地,作者也设计出一种P63/mmc Sc3MgH24三元相结构,该结构在200 GPa下相对于稳定的ScH6、MgH4和H2具有-0.54 meV/atom的形成能,该结构在常规的结构搜索设置下也同样无法被探寻。
该研究提出了一种理解金属超氢化合物结构稳定性的普适性原则。具有较强“模板效应”的金属晶格可有效降低氢共价网络的形成能,从而稳定住整体结构。借助这一理论,以具有较强“模板效应”的金属晶格为前驱结构,可有效提升搜索超导氢化物结构的效率,特别是目前第一性原理计算很难探索的三元及多元超氢化合物体系。作者们还在文章中指出,化学模板效应广泛存在于很多固体化合物之中。(来源:科学网)
相关论文信息:https://doi.org/10.1016/j.chempr.2022.10.015






