2021年12月6日,中科院昆明植物研究所杨玉荣团队在JACS《美国化学会志》上发表题为“Ir-Catalyzed Asymmetric Total Syntheses of Bisdehydrotuberostemonine D, Putative Bisdehydrotuberostemonine E and Structural Revision of the Latter”的新研究。
该研究针对百部碱合成中的难点多个连续手性中心的快速和精准控制并伴随复杂骨架的高效构建,发展了先进的铱催化不对称烯丙基化策略,把吡咯取代醇的Carreira不对称醛烯丙基化的立体发散合成与Krische反应一步手性合成5元内酯有机结合,为这些结构复杂百部碱提供了快捷高效的合成途径。
中科院昆明植物研究所博士生邓屹,副研究员梁箫、韦堃为该论文的第一作者;杨玉荣研究员为通讯作者。
百部碱(Stemona alkaloids)是从百部属植物中分离出的一类重要天然产物。这类生物碱常被用于治疗各种呼吸道疾病,例如百日咳,支气管炎和肺结核。百部生物碱因其具有重要生物活性和复杂的化学结构,尤其是在多环骨架中存在许多难以控制的手性中心,成为天然产物合成中具有很大挑战性目标分子。已有百部碱全合成研究文献中,消旋体的合成研究占多数,在为数不多的不对称合成中使用手性原料(通常是氨基酸)仍为常规手段,故发展催化不对称的合成新策略新方法对解决百部碱全合成具有重要意义。
Bisdehydrotuberostemonine型吡咯百部碱最早被发现于上世纪60年代,由于结构独特,除了具备百部碱多手性的合成难点外,在五环骨架中含有一个多取代的吡咯环,很大程度增加了合成挑战性。截至目前,关于此类bisdehydrotuberostemonine百部碱仅有美国著名化学家Wipf在2005年报道的一篇文献,从Cbz-L-络氨酸出发,需要28步,总产率0.3%才可完成其全合成。
鉴于上述难点,杨玉荣团队在前期运用铱催化不对称烯丙基化策略完成了许多复杂天然产物的研究经验上(如JACS. 2016, 138, 2560;ACIE. 2016, 55, 4044;JACS. 2017, 139, 3364;OL. 2017, 19, 6460;OL. 2018, 20, 4575;OL. 2019, 21, 8485;OL. 2021, 23, 2731;OL. 2021, 23, 7972等),创造性地设计了三次运用铱催化不对称烯丙基化的新策略巧妙完成bisdehydrotuberostemonine型吡咯百部碱的不对称全合成。如图1所示的逆合成分析,多环骨架中的两个5元内酯环相继通过Krische铱催化烯丙基化反应来依次构建,目标分子最终简化为一对双环化合物醛12a/b,即在C-9位置为非对映体化合物。为了快速高效并且能精准控制其手性中心,作者大胆设计了在吡咯取代醇的Carreira不对称醛烯丙基化的立体发散合成反应,希望能同时合成bisdehydrotuberostemonine D和E两个天然产物。特别指出:吡咯取代醇的Carreira不对称醛烯丙基化以及紧跟发生的Krische铱催化第二次烯丙基化反应,这样衔接自然、流线型的铱催化策略在天然产物合成中没有出现过。

图1:逆合成路线分析
作者从已知的5-甲酰基-1H-吡咯-2-羧酸甲酯15出发得到吡咯烯丙醇13,其和正丁醛的发生的Ir/amine双元催化Carreira烯丙基化反应以70%的产率以及>50:1 dr和99% ee获得关键中间体16a,或以65%的产率以及7:1 dr和99% ee获得非对映体16b。16a或16b 经过RCM反应顺利生成双环化合物12a或12b。
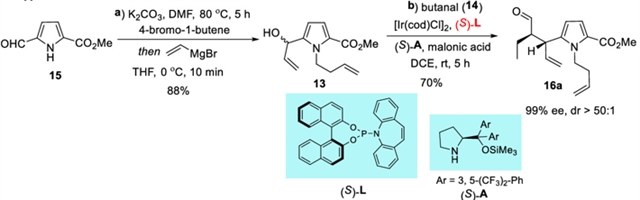
图2:铱/胺双元催化
接下来,作者使用Krische发展的条件,如图3所示,在催化剂(R)-Ir-tol-BINAP催化下12a仅用一步反应高效地得到手性γ-丁内酯18a,dr>20:1。18a具备了目标分子的绝大部分碳原子和关键的手性中心,作者发展的四步反应就能实现高效不对称合成,凸显出所设计的铱催化不对称烯丙基化新策略的强大威力。Ru催化将18a内酯环外双键异构化到环内得到11a。在尝试直接分子内环化成四环骨架不成功后,作者采用分步策略,先进行11a吡咯环碘代反应,随后再把生成的碘代物19a进行Heck反应得到四环骨架20a。氢化还原两个双键后立体选择性生成晶体化合物21a,利用单晶衍射确定化学结构后,采用Dibal-H还原内酯与吡咯取代的甲酯,再经过Ley-Griffith氧化得到吡咯醛10a。在对吡咯醛10a进行第三次铱催化不对称烯丙基化时,尽管产物22a收率不错,但却几乎是1:1的非对映体的混合物。经过对比研究,作者发现在吡咯醛为底物时,Krische烯丙基化产物容易发生γ-丁内酯的手性中心异构化。利用22a发生Takaya不对称氢化反应后,成功得到天然产物bisdehydrotuberostemonine D以及其18位的异构体。合成产物bisdehydrotuberostemonine D的化学结构得到了单晶衍射实验证实。有趣的是在与分离文献的核磁谱图认真比对后,作者发现18位的异构体的谱图与文献中推测9位异构体bisdehydrotuberostemonine E的谱图完全一致,即18位的异构体被误认为9位的异构体。

图3:百部碱bisdehydrotuberostemonine D合成路线
为了进一步澄清化合物bisdehydrotuberstemonine E的确切结构,作者对分离文献中推测的结构bisdehydrotuberostemonine E进行全合成。手性醛12b为起始物,经过和图3类似的步骤,推测的bisdehydrotuberostemonine E顺利被合成,该化合物经过单晶衍射实验证实。与预计一致,合成样品的核磁与分离作者的核磁谱图有明显差异,再次说明分离的bisdehydrotuberostemonine E结构鉴定错误,应该更正为bisdehydrotuberostemonine D的C18位的异构体,而非C9位异构体。为了和Wipf的路线比较,作者还合成了bisdehydrotuberostemonine,全部路线只需13步,该天然产物也第一次通过单晶衍射实验证实(如图4)。
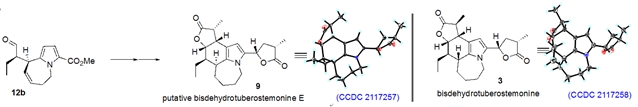
图4:百部碱putative bisdehydrotuberostemonine E和bisdehydrotuberostemonine
如图5所示,在文章的最后,作者对吡咯百部碱存在普遍性的结构不稳定进行了化学反应机理解释。

图5:C-18异构化过程
总之,在这项工作中,研究人员创造性地发展了铱催化不对称烯丙基化反应新策略,结合Ru和Pd过渡金属催化反应,全程未使用任何保护基,仅用12-13步简洁高效地完成了3个bisdehydrotuberostemonine型吡咯百部碱的不对称合成,为深入研究这类自然界含量低、生物活性有待研究的天然产物提供了高效获取的技术手段,发展的新策略将被扩展到更多天然产物的合成中。本研究中对bisdehydrotuberostemonine E结构更正对吡咯百部碱的植物化学研究也有重要意义。

图6:小结
该研究工作得到国家自然科学基金面上项目(21971249和22171274),中国科学院前沿局(QYZDB-SSW-SMC026和ZDBS-LY-SM030)的资助。(来源:科学网)
相关论文信息:https://doi.org/10.1021/jacs.1c11265






