|
|
|
|
|
QB | 前沿研究:传播性变异和新发变异的整合建模确定了先天性心脏病的新风险基因 |
|
|
论文标题:Integrative modeling of transmitted and de novo variants identifies novel risk genes for congenital heart disease (传播性变异和新发变异的整合建模确定了先天性心脏病的新风险基因)
期刊: Quantitative Biology
作者:Mo Li, Xue Zeng, Chentian Jin, Sheng Chih Jin, Weilai Dong, Martina Brueckner, Richard Lifton, Qiongshi Lu, Hongyu Zhao
发表时间:27 May 2021
DOI:10.15302/J-QB-021-0248
微信链接:点击此处阅读微信文章
全外显子组测序 (WES) 研究已经确定了先天性心脏病 (CHD) 先证者中富含新发突变 (DNM) 的多个基因。然而,由于 CHD 的异质性病因和每个基因的低突变率,仅基于 DNM 的风险基因识别在统计学上仍然具有挑战性。近日, 来自美国耶鲁大学的赵宏宇教授研究组开发了一个联合分析新发变异和罕见传播变异的建模框架,即TADA-R框架。该框架是一种创新的统计检验,通过联合分析新发和罕见的传播变异来识别性状相关基因。本文将此方法应用于2,645个CHD先证者-父母三人组的 WES 数据,确定了15个重要的基因,其中许多是新的风险基因,这些发现为CHD的遗传病学提供了重要线索。相关论文 “Integrative modeling of transmitted and de novo variants identifies novel risk genes for congenital heart disease” (点击文末“阅读原文”下载PDF全文)发表在Quantitative Biology期刊上。

全文概要
先天性心脏病 (CHD) 是一种常见的出生缺陷,影响 0.8% 的活产儿。众所周知,遗传和环境因素在 CHD 中都发挥着重要作用,一项针对丹麦双胞胎的研究估计该人群的遗传遗传力接近 0.5。然而,对 CHD 的遗传基础缺乏全面了解是对 CHD 患者进行生殖咨询的主要障碍。在孟德尔疾病和一些异质性单基因疾病, 如听力损失和复杂的疾病如心血管疾病中, WES研究已经成功地确定了一些新的致病基因。由于WES经常在每个外显子组中发现数以万计的遗传变异,其中大部分与感兴趣的疾病无关,研究人员需要缩小所考虑的变异库。例如,对于健康父母及其受影响后代的外显子组测序,只有那些在普通人群中频率极低的变体或儿童中的新发突变(DNMs)才会被进一步研究,从而大大减少考虑的变体数量。
由于突变数量少,对DNMs的研究往往缺乏统计能力。每个外显子组估计有1.2个DNMs,因此,即使在特定基因导致疾病表型的情况下,病例和对照组中DNMs的数量差异通常也很小。传播和新发关联(TADA)框架是一种层次化的贝叶斯方法,通过从外显子组的遗传和新发变异中获取信息来识别疾病基因。尽管取得了一些成功,但TADA在方法上有一些限制。它没有纳入隐性遗传模式,也没有考虑影响每个基因从头变异的因素(如局部序列背景)。
在本项工作中,作者介绍了TADA-R,这是一个建立在TADA基础上的通用模型,包括隐性疾病模型,即子代具有隐性基因型,包括同型杂合子 (即患儿从父母双方各继承了两个相同的突变),以及复合杂合子 (即患儿继承了同一基因的两个不同突变,从父母各继承一个)。通过考虑显性效应和隐性效应,该模型对不同的遗传结构都有适应性。该模型还结合了基因层面的注释(如基因长度和序列背景)和来自人群参考板(如gnomAD)的数据,以更准确地描述新发和传播的罕见变体的预期频率,这进一步提高了方法的统计能力。作者将TADA-R模型应用于2,645个CHD亲属三人组的WES数据, 总共发现了15个重要的基因,其中许多是新的。这些发现为CHD的遗传病学提供了重要线索。
首先作者介绍了TADA-R模型的基本结构(如图1), TADA-R 模型的关键是亲子三人组中基因发生罕见有害突变的概率。

图1. 量化基因-疾病关联程度的检验统计量
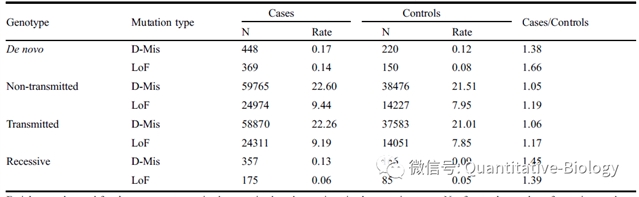
作者在测序数据上运行了发现变异的方法, 发现与对照组相比,CHD病例三人组中富含新的缺失性(D-Mis)和功能丧失性(LoF)突变, 而病例中的可容忍错义(T-Mis)和同义DNMs没有富集(表1)。这证实,与健康对照组相比,CHD患者携带更多的破坏性、改变蛋白质的DNM。因此,在下面的分析中只考虑LoF和D-Mis突变。
表1. 2645个CHD三人组相比对照组的突变计数
然后作者在模拟数据上评估了该方法的I型错误和统计功效。作者比较了四种方法:(1)TADA-Denovo,(2)TADA,(3)TADA-R,和(4)TADA-R与特定基因的先验(图2)。在所有的模拟设置中,具有特定基因先验的TADA-R具有最好的性能, 而且纳入特定基因的先验进一步提高了统计能力。与TADA相比,没有/有特定基因先验的TADA-R的统计能力提高了15.6%和25.4%。
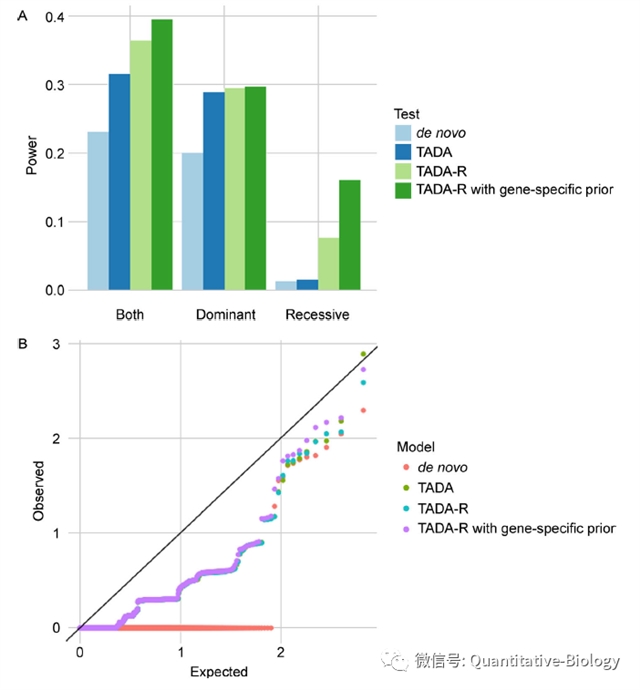
图2. 不同模型的统计功效和I型错误
最后,作者对2,645个CHD患者的亲属三人组进行了TADA-R分析。15个基因达到了全基因组的显著水平(如表2)。
表2. CHD患者-亲属三人组TADA-R分析鉴定得到的显著差异基因

此外,作者还将这些显著基因的贝叶斯因子分解为显性三重奏(即新发、非传播和传播三重奏)和隐性三重奏的贡献(如图3)。结果发现其中的11个基因(CHD7, KMT2D, PTPN11, RBFOX2, POGZ, ACTB, CYP21A2, RPL5, AKAP12, NOTCH1 和SMAD2)只显示显性遗传, 8个在关联分析或基因表达分析中被报道为人类CHD基因, 4个基因(GDF1, SULF1, NSD1和ADIPOQ)同时显示出显性和隐性关联。

图3. TADA-R发现的显著基因的贝叶斯系数、pLI得分和发育小鼠心脏中的基因表达量
最后, 作者总结了TADA-R方法可能的局限性。尽管在分析中包括传播性变异可能会提高统计能力,但它可能会给解释结果带来困难。在本文的分析中,作者将突变的贡献分解为显性效应和隐性效应,这使作者更容易解释疾病机制, 然而,还需要有进一步分解的方法。另一个方面是改模型缺乏对近亲繁殖的考虑, 对病例中近亲繁殖率的提高进行建模,有可能进一步提高模型的性能。除了CHD,TADA-R还可以应用于分析其他疾病的WES数据。随着更多性状和更多个体的WES数据的不断产生,作者希望TADA-R能带来更多的基因定型和生物学发现。
摘要
Background: Whole-exome sequencing (WES) studies have identified multiple genes enriched for de novo mutations (DNMs) in congenital heart disease (CHD) probands. However, risk gene identification based on DNMs alone remains statistically challenging due to heterogenous etiology of CHD and low mutation rate in each gene.
Methods: In this manuscript, we introduce a hierarchical Bayesian framework for gene-level association test which jointly analyzes de novo and rare transmitted variants. Through integrative modeling of multiple types of genetic variants, gene-level annotations, and reference data from large population cohorts, our method accurately characterizes the expected frequencies of both de novo and transmitted variants and shows improved statistical power compared to analyses based on DNMs only.
Results: Applied to WES data of 2,645 CHD proband-parent trios, our method identified 15 significant genes, half of which are novel, leading to new insights into the genetic bases of CHD.
Conclusion: These results showcase the power of integrative analysis of transmitted and de novo variants for disease gene discovery.
Quantitative Biology期刊介绍
Quantitative Biology (QB)期刊是由清华大学、北京大学、高教出版社联合创办的全英文学术期刊。QB主要刊登生物信息学、计算生物学、系统生物学、理论生物学和合成生物学的最新研究成果和前沿进展,并为生命科学与计算机、数学、物理等交叉研究领域打造一个学术水平高、可读性强、具有全球影响力的交叉学科期刊品牌。
《前沿》系列英文学术期刊
由教育部主管、高等教育出版社主办的《前沿》(Frontiers)系列英文学术期刊,于2006年正式创刊,以网络版和印刷版向全球发行。系列期刊包括基础科学、生命科学、工程技术和人文社会科学四个主题,是我国覆盖学科最广泛的英文学术期刊群,其中13种被SCI收录,其他也被A&HCI、Ei、MEDLINE或相应学科国际权威检索系统收录,具有一定的国际学术影响力。系列期刊采用在线优先出版方式,保证文章以最快速度发表。
高等教育出版社入选“中国科技期刊卓越行动计划”集群化项目。Frontier系列期刊中:13种被SCI收录;1种被A&HCI收录;6种被Ei收录;2种被MEDLINE收录;11种中国科技核心期刊;16种被CSCD收录。
中国学术前沿期刊网
http://journal.hep.com.cn

特别声明:本文转载仅仅是出于传播信息的需要,并不意味着代表本网站观点或证实其内容的真实性;如其他媒体、网站或个人从本网站转载使用,须保留本网站注明的“来源”,并自负版权等法律责任;作者如果不希望被转载或者联系转载稿费等事宜,请与我们接洽。






