|
|
|
|
|
中美科学家合作实现吡啶C3-H直接烯基化反应新进展 |
|
|
2021年10月11日,南开大学叶萌春/Scripps研究所余金权团队合作在Nature Chemistry上发表了一篇题为“A Directive Ni Catalyst Overrides Conventional Site-Selectivity in Pyridine C–H Alkenylation”的研究工作。通过引入功能化卡宾配体协同的镍-铝(Ni(0)-Al(III))双金属催化策略,该团队实现了一当量吡啶C3-H的直接烯基化反应。博士生张涛为论文第一作者,南开大学为第一完成单位。
吡啶衍生物广泛存在于医药、农药和具有生物活性的天然产物中:目前已知有7000种以上的生物活性分子含有吡啶结构,全球畅销药物的前200强中约有20%的药物是吡啶衍生物。因此,发展原子和步骤经济性吡啶衍生物的合成方法,对于构建吡啶衍生物具有重要的研究意义。相比于传统的官能化方法,过渡金属催化的吡啶C-H直接官能化方法受到了最为广泛的关注,因为其不仅具有较高的原子和步骤经济性,而且具有原料来源广泛价廉易得的特点。然而由于吡啶骨架通常含有多个C-H位点,以及吡啶环的缺电性和N原子的强配位性,吡啶C-H选择性官能化反应是领域内一个非常大的挑战。其中,C2-H和C4-H最为缺电子,相对容易通过氧化加成的方式活化,但是C3-H缺电性稍弱,既不易通过氧化加成的方式,也不易通过亲电活化的方式活化。因此,尽管C3-烯化的吡啶衍生物具有重要的生物活性,但是如何通过吡啶C-H烯化方法合成此类化合物一直是个长期的挑战。

2011年余金权课题组发现强配位的双齿配体可以抑制吡啶对Pd(II)的配位失活,首次实现了吡啶C3-H的烯基化反应(J. Am. Chem. Soc. 2011, 133, 19090)。尽管较好的控制了C3位选择性,但是该方法需要使用大大过量的吡啶(至少16 equiv),且需要足够的吡啶浓度才能保证合适的反应活性。这个局限性导致该方法仅适用于简单吡啶,无法兼容广泛存在的具有生物活性的复杂吡啶分子。因此,如何发展一当量吡啶参与的高效C3-烯化反应受到了持续的关注和广泛的研究,但是一直难以突破。
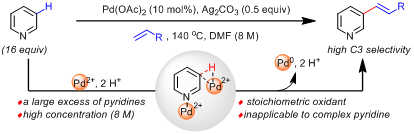
为了解决这一难题,该团队借鉴了吡啶C2-H和C4-H官能化的成功经验:Nakao/Hiyama和Ong课题组分别于2008年和2010年实现了低价金属Ni(0)催化的吡啶C2-H和C4-H的直接烯基化反应(J. Am. Chem. Soc. 2008, 130, 2448 & J. Am. Chem. Soc. 2010, 132, 11887)。反应中,合适的Lewis acid被用来加强吡啶的缺电性,从而更容易促进C2-H和C4-H和低价金属Ni(0)的氧化加成,进而通过配体位阻的控制,可以顺利实现一当量吡啶的C2-H和C4-H的选择性烯化反应。

鉴于这一成功经验,该团队巧妙地设想在两类金属中间引入合适长度和位阻的配体:这样通过位阻调节,抑制C2-H和金属的氧化加成;同时通过配体的张力效应,限制金属Ni到达C4-H,从而最终只能活化C3-H。此外,由于Lewis acid的使用,该方法在仅使用一当量吡啶条件下应该仍然具有较好的反应活性,从而同时解决活性和C3-选择性的难题。

经过一系列系统的配体优化和条件筛选,他们发现使用羟乙基修饰的卡宾配体,可以协同Ni-Al双金属实现吡啶C3-H的选择性烯基化。在优化的反应条件下,多种含有生物活性结构的吡啶衍生物均能够以43-89%的收率和高达97:3的位点选择性,实现吡啶C-H后期烯基化修饰。
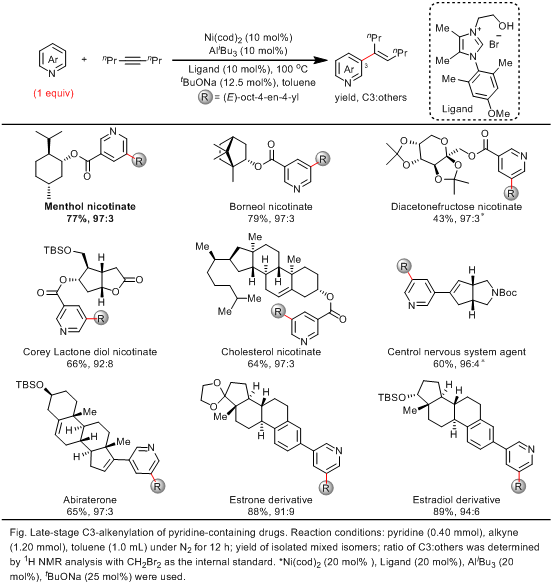
叶萌春/余金权团队发展了新型的双官能化卡宾配体,通过卡宾配体协同的镍-铝(Ni(0)-Al(III))双金属催化策略,实现了一当量吡啶的C3-H烯基化反应。该方法为广泛存在的生物活性吡啶结构分子的后期修饰提供了一条经济和高效的途径。此外,配体协同的双金属催化剂实现C-H非导向的远程选择性控制的方法有望在其他底物和金属催化的反应中得到广泛的应用。(来源:科学网)
相关论文信息:https://doi.org/10.1038/s41557-021-00792-1






