|
|
|
|
|
面包小麦精细表观组图谱绘制及全基因组顺式作用元件鉴定 | Genome Biology |
|
|
论文标题:The bread wheat epigenomic map reveals distinct chromatin architectural and evolutionary features of functional genetic elements
期刊:Genome Biology
作者:Zijuan Li, Meiyue Wang, Kande Lin, Yilin Xie, Jingyu Guo, Luhuan Ye, Yili Zhuang, Wan Teng, Xiaojuan Ran, Yiping Tong, Yongbiao Xue, Wenli Zhang and Yijing Zhang
发表时间:2019/07/15
DOI:10.1186/s13059-019-1746-8
微信链接:https://mp.weixin.qq.com/s/ZhtRdOb62c36FDX9zfBRmA
面包小麦(Triticum aestivum)是世界上产量第二高的谷物,全球种植面积超过2.15亿公顷,是人类食物中植物蛋白的主要来源。深入解析小麦功能基因表达调控元件及调控网络,将助力于精准分子辅助育种,加快优质高产多抗小麦的种质创新进程。
近日,中国科学院植物生理生态研究所和南京农业大学团队合作生成并绘制面包小麦精细的表观组图谱,以此为基础针对性开发整合计算流程对全基因组顺式调控元件进行了系统的挖掘与鉴定,并初步探索了其作用机制,为小麦基因调控机制解析研究提供了重要的资源。该成果发表在Genome Biology 杂志。

面包小麦是一种异源六倍体,有3组7对染色体,分别来自于3个不同的祖先种(Triticum uratru,Aegilops speltoides 以及Aegilops tauschii)。高质量的面包小麦全基因组序列于2019年初公布,大小约为16 Gb,是人类基因组的5倍。其中93%是非编码序列,蕴含着丰富的基因远端调控元件,在小麦全基因组水平准确鉴定顺式元件并解析其调控机制,是研究小麦多倍化及驯化过程中基因表达调控的关键步骤。由于表观修饰在基因调控过程中发挥了重要作用,有机整合表观组信息有助于在全基因组水平精准预测顺式调控区域。但是,与基因组序列相对简单的模式生物相比,庞大而复杂的小麦基因组对组学数据产生与数据分析及机制解析均带来巨大挑战。
合作团队生成并整合数十套小麦表观组数据,通过开发和运用针对小麦表观组数据的整合计算方法与分析流程,系统挖掘了小麦的全基因组顺式调控元件并初步探索其作用机制,为小麦基因调控机制解析研究提供了重要的资源,主要包括以下方面:
1)刻画不同类型调控元件的表观修饰组合模式,以此为基础预测上千个新的顺式作用元件,其中包含启动子和增强子等。通过瞬时转化实验进一步证明功能元件预测具有较高的准确度。上述工作锁定六倍体小麦中1.5%基因组区域存在活跃的顺式元件,为小麦功能基因组研究提供了重要的参考信息,极大程度减少基因调控机制研究的工作量。
2)鉴定增强子与启动子的特异性序列特征,并揭示二者的差异调控机制。
3)通过对小麦3套亚基因组的比较揭示了基因调控的选择压力作用于序列和染色质结构两个层面。
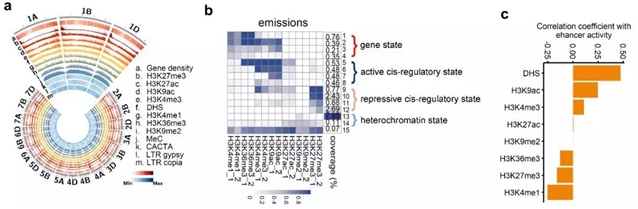
刻画面包小麦表观组图谱(a)并对全基因组顺式作用元件进行挖掘(b)与活性鉴定(c)
该研究工作由中国科学院植物生理生态研究所,南京农业大学和中国科学院遗传与发育生物学研究所团队合作完成。中国科学院植物生理生态研究所的张一婧研究员和南京农业大学的张文利教授为论文的共同通讯作者;中国科学院遗传与发育生物学研究所的薛勇彪研究员与童依平研究员参与项目的设计与指导,博士生李子娟、王梅月、林堪德、谢忆琳为共同第一作者。该研究受到中科院战略科技先导专项、基金委和教育部项目的资助。
摘要:
Background
Bread wheat is an allohexaploid species with a 16-Gb genome that has large intergenic regions, which presents a big challenge for pinpointing regulatory elements and further revealing the transcriptional regulatory mechanisms. Chromatin profiling to characterize the combinatorial patterns of chromatin signatures is a powerful means to detect functional elements and clarify regulatory activities in human studies.
Results
In the present study, through comprehensive analyses of the open chromatin, DNA methylome, seven major chromatin marks, and transcriptomic data generated for seedlings of allohexaploid wheat, we detected distinct chromatin architectural features surrounding various functional elements, including genes, promoters, enhancer-like elements, and transposons. Thousands of new genic regions and cis-regulatory elements are identified based on the combinatorial pattern of chromatin features. Roughly 1.5% of the genome encodes a subset of active regulatory elements, including promoters and enhancer-like elements, which are characterized by a high degree of chromatin openness and histone acetylation, an abundance of CpG islands, and low DNA methylation levels. A comparison across sub-genomes reveals that evolutionary selection on gene regulation is targeted at the sequence and chromatin feature levels. The divergent enrichment of cis-elements between enhancer-like sequences and promoters implies these functional elements are targeted by different transcription factors.
Conclusions
We herein present a systematic epigenomic map for the annotation of cis-regulatory elements in the bread wheat genome, which provides new insights into the connections between chromatin modifications and cis-regulatory activities in allohexaploid wheat.
(来源:科学网)
特别声明:本文转载仅仅是出于传播信息的需要,并不意味着代表本网站观点或证实其内容的真实性;如其他媒体、网站或个人从本网站转载使用,须保留本网站注明的“来源”,并自负版权等法律责任;作者如果不希望被转载或者联系转载稿费等事宜,请与我们接洽。






