|
|
|
|
|
人类基因组的病毒基因含量比我们预想的更为多变 | Mobile DNA |
|
|
论文标题:Variation in proviral content among human genomes mediated by LTR recombination
期刊:Mobile DNA
作者:Jainy Thomas, Hervé Perron and Cédric Feschotte
发表时间:2018/12/18
数字识别码:10.1186/s13100-018-0142-3
原文链接:https://mobilednajournal.biomedcentral.com/articles/10.1186/s13100-018-0142-3?utm_source=other&utm_
medium=other&utm_content=null&utm_campaign=BSCN_2_WX_mobilednajournal_arti_scinet
微信链接:https://mp.weixin.qq.com/s/6-a39RauaRAgFg7tS7oXkg
人类DNA的一部分其实来源于病毒,其中许多DNA是在数百万年前被插入到人类祖先的原始遗传物质中,并且自此之后一直被后代遗传。因此,人们认为这些物质在现代人类的基因组中变化不大。人内源性逆转录病毒(HERV)是迄今为止人类基因组中最常见的病毒衍生序列。在Mobile DNA 上发表的一项最新研究展示了一种机制,这种机制在人与人之间引入了较之前预期更多的HERV含量个体差异。

人类DNA中有一部分是来源于病毒,其中许多DNA是在数百万年前被插入到人类祖先的原始遗传物质中,并一直被后代遗传。人内源性逆转录病毒(HERV)是迄今为止人类基因组中最常见的病毒衍生序列。大多数HERV序列早已被同化,因此在人群中的所有个体中共存。但并非所有HERV序列都如此,已发现有少数HERV序列仅存在于部分个体中。已知这些未固定的HERV元件中的大多数是来自相对近期的插入事件,这些插入事件在人群中仍处于分离状态。但最近在Mobile DNA 上发表的一项新研究表明,另一种机制已在人与人之间引入了较之前预期更多的HERV含量个体差异。为何会如此呢?
首先,考虑HERV的结构特征是很重要的。为了整合到宿主染色体中,这些序列必须是称为前病毒的全长元件。每个前病毒围绕中心核组织,该中心核含有夹在每个末端重复的长链非编码序列之间的病毒编码基因,称为长末端重复序列(LTRs)(参见图1)。整合后,前病毒的两个LTR(在插入时相同)频繁重组以形成所谓的solo LTR。重组过程中消除了内部病毒基因以及两个LTR中的一个,留下单个LTR。之前已经估算出人类基因组中90%的HERV是单个LTR,并且只有10%保持其前病毒形式。但如果这些前病毒元件中的一些仍在转变为单个LTR会怎样?犹他大学(University of Utah)和康奈尔大学(Cornell University)的研究人员着手研究了这一问题,并评估了LTR重组过程在人类中产生HERV变异的程度。
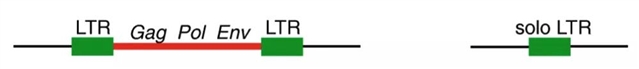
图2. 典型前病毒的结构,其内部区域(红线)编码gag、pol和env基因,两侧为两个长末端重复序列(LTR)。异位重组发生在前病毒的两个LTR之间,导致内部区域连同一个LTR一起缺失,从而形成单个LTR。
Jainy Thomas博士开发了一种新的计算方法,这种方法使得他们能够筛选来自不同人群的大量DNA序列,以发现可能的罕见LTR重组事件。鉴于人类基因组中存在大量的HERV序列,这项任务犹如大海捞针。研究者从一个由Simons基金会支持的、包含130个不同遗传群体的全基因组序列的公开数据集中搜索三个逆转录病毒家族的变异体:HERV-K(HML2)、HERV-W和HERV-H。Thomas博士开发的方法允许其找到之前登记的大部分HERV变异体,并发现更多的变异体(图2)。不足为奇的是,大多数新发现的变异体显然非常罕见,因为其仅在一个或几个个体中发现。但其也是出乎意料的,因为这些HERVs中有许多早已插入人类祖先的DNA中,有些甚至与亲缘关系最近的类人猿共享,因此被认为是固定在人类群体中的。尽管如此,Thomas博士能够通过实验证实在人群中确实分离出这些变异体中的几种,从而验证了她的计算方法的有效性。
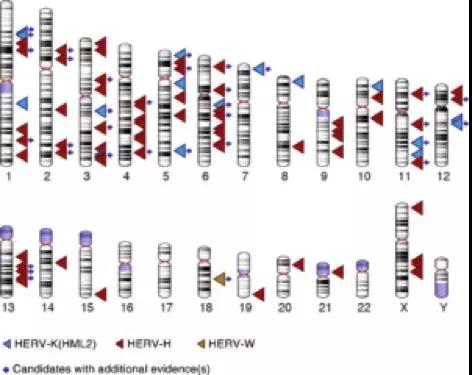
图3. 候选二态HERV位置的核型图
这种类型的HERV变异体是否具有重要意义?有许多理由可以认为,这些遗传变异可能是导致疾病易感性的人类生理变异的一个被忽视的来源。事实上,有越来越多的证据表明,研究中调查的三个HERV家族对人类同时发挥有益和致病作用。例如,HERV编码的基因在罹患肌萎缩性脊髓侧索硬化症(ALS或Lou Gehrigs病)、多发性硬化症和一些癌症等疾病的个体中过表达,其基因产物被认为与这些疾病的病因或进展有关。另一方面,一些HERV似乎具有有益的特性。最近,Feschotte博士及其同事的研究表明,一些HERV对于人体免疫反应的适当调节至关重要。前病毒HERV-H元件是新近发表在Mobile DNA 上的研究中揭示的变异来源之一,已证实其对保持胚胎细胞的多能性(即分化为不同细胞类型的能力)非常重要。因此,更好地量化这些前病毒HERV在个体和群体中的存在或缺失,对于更好地理解这些元件如何影响人类的健康和生理机能将非常重要。
摘要:
Background
Human endogenous retroviruses (HERVs) occupy a substantial fraction of the genome and impact cellular function with both beneficial and deleterious consequences. The vast majority of HERV sequences descend from ancient retroviral families no longer capable of infection or genomic propagation. In fact, most are no longer represented by full-length proviruses but by solitary long terminal repeats (solo LTRs) that arose via non-allelic recombination events between the two LTRs of a proviral insertion. Because LTR-LTR recombination events may occur long after proviral insertion but are challenging to detect in resequencing data, we hypothesize that this mechanism is a source of genomic variation in the human population that remains vastly underestimated.
Results
We developed a computational pipeline specifically designed to capture dimorphic proviral/solo HERV allelic variants from short-read genome sequencing data. When applied to 279 individuals sequenced as part of the Simons Genome Diversity Project, the pipeline retrieves most of the dimorphic loci previously reported for the HERV-K(HML2) subfamily as well as dozens of additional candidates, including members of the HERV-H and HERV-W families previously involved in human development and disease. We experimentally validate several of these newly discovered dimorphisms, including the first reported instance of an unfixed HERV-W provirus and an HERV-H locus driving a transcript (ESRG) implicated in the maintenance of embryonic stem cell pluripotency.
Conclusions
Our findings indicate that human proviral content exhibit more extensive interindividual variation than previously recognized, which has important bearings for deciphering the contribution of HERVs to human physiology and disease. Because LTR retroelements and LTR recombination are ubiquitous in eukaryotes, our computational pipeline should facilitate the mapping of this type of genomic variation for a wide range of organisms.
阅读论文全文请访问:
https://mobilednajournal.biomedcentral.com/articles/10.1186/s13100-018-0142-3?utm_source=other&utm_
medium=other&utm_content=null&utm_campaign=BSCN_2_WX_mobilednajournal_arti_scinet
期刊介绍:
Mobile DNA(https://mobilednajournal.biomedcentral.com/,5.891 - 2-year Impact Factor, 3.957 - 5-year Impact Factor) is an open access, peer-reviewed journal that publishes articles providing novel insights into DNA rearrangements in all organisms, ranging from transposition and other types of recombination mechanisms to patterns and processes of mobile element and host genome evolution. In addition, the journal will consider articles on the utility of mobile genetic elements in biotechnological methods and protocols.
(来源:科学网)
特别声明:本文转载仅仅是出于传播信息的需要,并不意味着代表本网站观点或证实其内容的真实性;如其他媒体、网站或个人从本网站转载使用,须保留本网站注明的“来源”,并自负版权等法律责任;作者如果不希望被转载或者联系转载稿费等事宜,请与我们接洽。






