复旦大学公共卫生学院教授粟硕团队与中国科学院微生物研究所研究员毕玉海等合作者,揭示了多种哺乳动物宏基因组数据中的病毒基因组组成、生态学特征与潜在跨物种传播风险,为构建“人-动物-环境”一体化、多维度新发传染病预测预警体系奠定了重要数据基础。9月4日,相关研究发表于《自然》。
21世纪以来,人类多次经历由动物源新发传染病原引发的大流行疫情。据报道,超过70%的人畜共患疾病源自野生动物。同时国内外大量研究提示,部分养殖哺乳动物如水貂等可能具有携带人兽共患病毒的潜力。然而除传统家畜外,养殖哺乳动物的病原生态学与流行病学数据存在较大空白,制约了新发传染病疫情的精准防控。
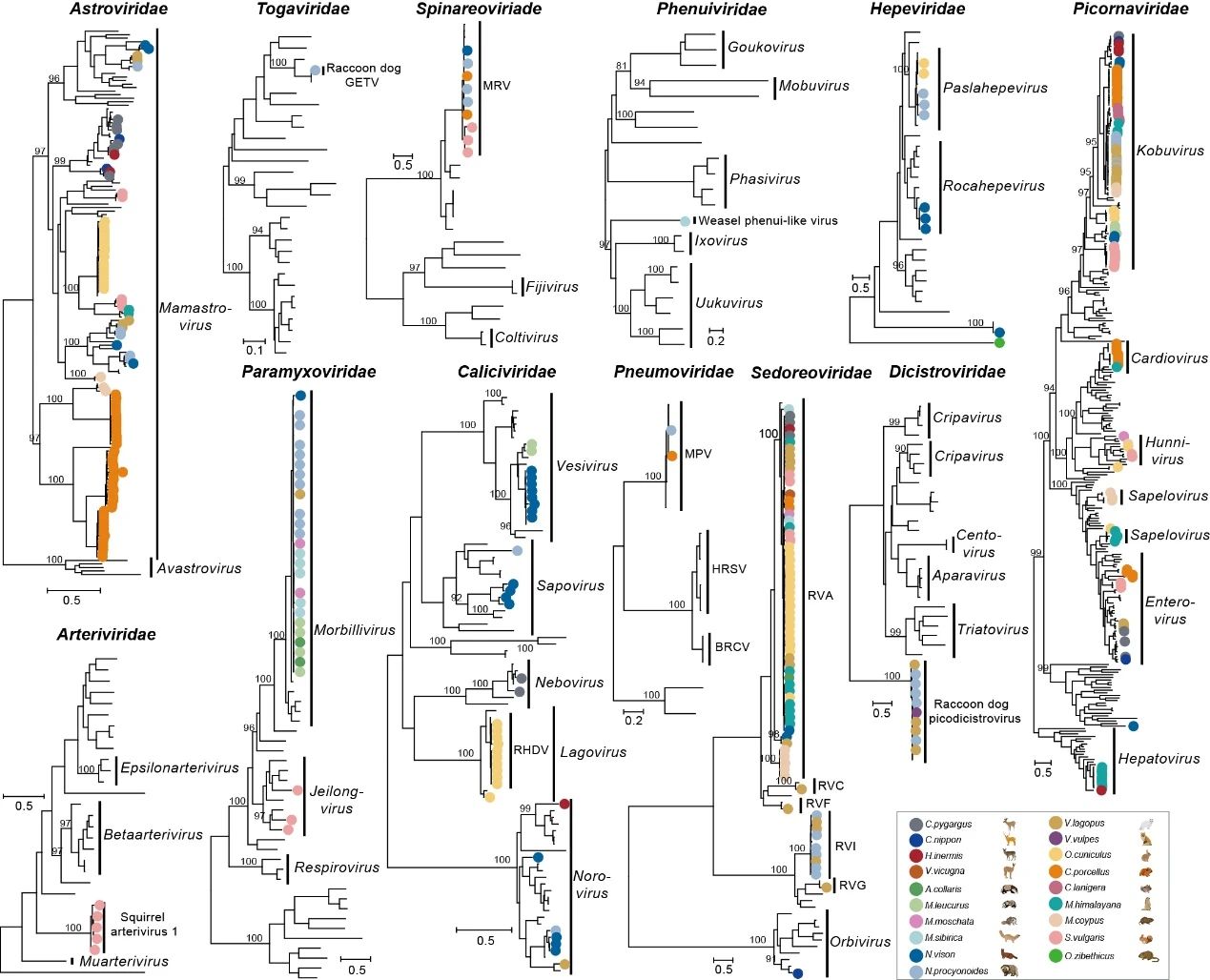
研究团队对5个动物目的哺乳动物进行了系统的宏基因组研究,鉴定到125种病毒基因组,其中39种可推定为新的病毒种。进一步地,研究人员遴选出诺如病毒、盖塔病毒等多种频繁发生“宿主跳跃”的潜在“风险”病毒,对其潜在跨物种传播风险进行解析,并从空间聚类、动物类群、种群、组织等维度揭示了这些病毒的生态学与流行病学特征。
哺乳动物携带RNA病毒的多样性。图片来源于《自然》
为在个体水平解析病毒共感染,同时更灵敏地挖掘低丰度病毒,团队设计了单组织单样本的建库策略,在166个单组织文库里鉴定到2种以上病毒。结果表明,星状病毒、细小病毒等与其他病毒频繁发生共感染。此外,研究人员对低丰度病毒片段进行精准拼接,避免了真阳性病毒因丰度阈值而被过滤,实现了多个病毒基因组90%的完成度。
“我们希望填补当前对动物携带微生物的认知空白,尤其是养殖哺乳动物。”粟硕表示,“如果能提前监测和预报对人类和家畜构成潜在威胁的病毒,并部署包括动物疫苗研发在内的综合防控措施,将对于新发传染病关口前移、保障生命健康至关重要。”
相关论文信息:https://doi.org/10.1038/s41586-024-07901-3
版权声明:凡本网注明“来源:中国科学报、科学网、科学新闻杂志”的所有作品,网站转载,请在正文上方注明来源和作者,且不得对内容作实质性改动;微信公众号、头条号等新媒体平台,转载请联系授权。邮箱:shouquan@stimes.cn。