|
|
|
|
|
测序结果多如山,用作研究背景分析可好?| Microbiome |
|
|
论文标题:Culture-enriched human gut microbiomes reveal core and accessory resistance genes
期刊:Microbiome
作者: Frédéric Raymond,Maurice Boissinot, Amin Ahmed Ouameur,Maxime Déraspe,Pier-Luc Plante,Sewagnouin Rogia Kpanou,Ève Bérubé, Ann Huletsky, Paul H. Roy, Marc Ouellette, Michel G. Bergeron, Jacques Corbeil
发表时间:2019/04/05
数字识别码: 10.1186/s40168-019-0669-7
原文链接:http://t.cn/E9mDQIR
近期在Microbiome发表了新文章的作者与我们分享了自己研究背后的故事:“在分析基因组数据时,我通常喜欢物尽其用,尽可能充分利用每种方法。我们最近发表的论文主要研究了肠道微生物,特别是低丰度细菌中的抗生素抗性基因。在这里,我想和大家分享一下研究所用方法背后的故事”。

图1
我们最近在Microbiome上发表的论文是我的“抗生素对微生物组影响三部曲”中的第三篇。在三部曲的第一篇(The ISME Journal , 2016)中,我们确定了摄入某一常用抗生素是如何对粪便微生物组产生影响的。这项研究的亮点之一是我们使用了深度无选择性宏基因组学分析(每个样本中的基因数量高达15 Gb);这种分析方法不仅可以对微生物组进行分类分析,还可以进一步挖掘宏基因组中的抗生素抗性基因。 我们最终检测到了43000多个可能的抗生素抗性基因!
的确,43000是一个非常庞大的数字。这意味着仅在24个健康的人(每个对象分别在3个时间点取样)中就存在着43000多个可能对抗生素存在抗性的细菌。当时看到这个结果,我整个人都不好了!但现在来看的话,这个数字却有着完全不同的启示。
在这篇文章(以及第二篇文章)发表几个月后,我有幸前往法国阿讷西参加Mérieux基金会和巴斯德研究所联合举办的为期两周的抗生素进阶课程。课程内容主要围绕抗生素及抗生素耐药性,授课老师包括多个领域的十几位专家。在这两周的时光中,不单单是授课和学习,也可以说是一种人文经历。课程期间我被邀请简要介绍抗生素抗性和微生物组的研究进展,我分享了那篇2016年ISME论文的未发表数据,这些数据揭示了某些特定的抗生素抗性基因普遍存在于我们的微生物组中。当时一位很有名的微生物学家举手说到:“这些抗生素抗性基因中许多存在于所有的大肠杆菌菌株中。它们与临床不相关。它们无处不在!”
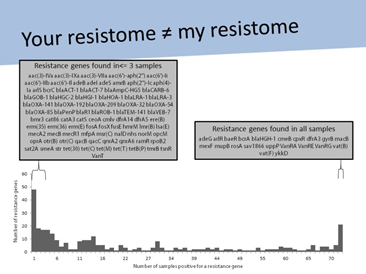
图2:我们的耐药基因组不尽相同
我反复思考这句话并逐渐意识到,对于这些可能的抗生素抗性基因,我们要做的不仅仅是归类。43000这个庞大的数字只是一个纸老虎,它没有任何意义。我们需要把 这些基因放在特定背景中进行研究:基因组学背景、流行病学背景、临床医学背景。
在接下来的一年里,我和同事们试图找到从大数据角度进行比较基因组学研究的方法(例如使用Ray Surveyor软件)。方法学研究日臻完善的同时我又开始思考该如何利用团队在方面的专长对公共数据库中可用的基因组数据进行分析解读以更深入全面地了解微生物的基因情况。以下就是我们逐步推进的研究:
• 对含有抗生素抗性基因的重叠群进行了分类
• 对某种细菌(例如大肠杆菌)所有可获得的基因组数据中的抗生素抗性基因进行了标注
• 将在可能源自大肠杆菌的重叠群中发现的抗生素抗性基因与实际在大肠杆菌基因组中发现的抗性基因进行比较
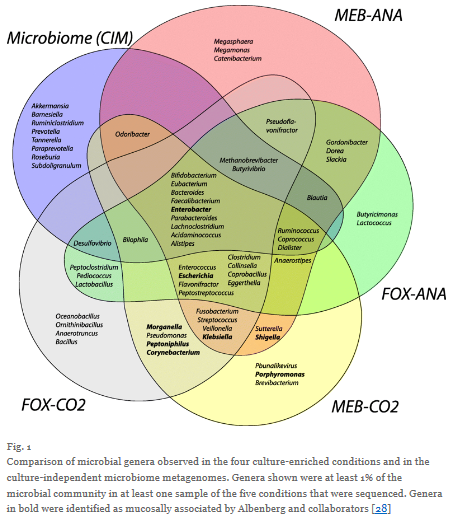
图3
在这里我就不再对分析结果加以赘述了(如果您感兴趣,请点击此处,阅读完整论文),但是这些分析方法的确帮助我们确定了哪些抗生素抗性基因可能具有实际意义,哪些基因最可能发生水平转移。至于具体的实现方式,我们主要考量了这些基因的基因组环境以及它们在不同隔离种群中的分布。
总之,我想强调的重点只有一个,即研究背景。现在有如此之多的测序结果可以为我们的数据提供研究背景分析,任由这些结果荒废而不加以利用简直是愚不可及。坐拥海量数据,真正限制我们的只是我们的想象力,抑或是计算能力……
摘要:Low-abundance microorganisms of the gut microbiome are often referred to as a reservoir for antibiotic resistance genes. Unfortunately, these less-abundant bacteria can be overlooked by deep shotgun sequencing. In addition, it is a challenge to associate the presence of resistance genes with their risk of acquisition by pathogens. In this study, we used liquid culture enrichment of stools to assemble the genome of lower-abundance bacteria from fecal samples. We then investigated the gene content recovered from these culture-enriched and culture-independent metagenomes in relation with their taxonomic origin, specifically antibiotic resistance genes. We finally used a pangenome approach to associate resistance genes with the core or accessory genome of Enterobacteriaceae and inferred their propensity to horizontal gene transfer.
阅读论文全文请访问:http://t.cn/E9mDQIR
期刊介绍:Topics broadly addressing the study of microbial communities, such as, microbial surveys, bioinformatics, meta-omics approaches and community/host interaction modeling will be considered for publication. Through this collection of literature Microbiome hopes to integrate researchers with common scientific objectives across a broad cross-section of sub-disciplines within microbial ecology.
(来源:科学网)
特别声明:本文转载仅仅是出于传播信息的需要,并不意味着代表本网站观点或证实其内容的真实性;如其他媒体、网站或个人从本网站转载使用,须保留本网站注明的“来源”,并自负版权等法律责任;作者如果不希望被转载或者联系转载稿费等事宜,请与我们接洽。






