导言
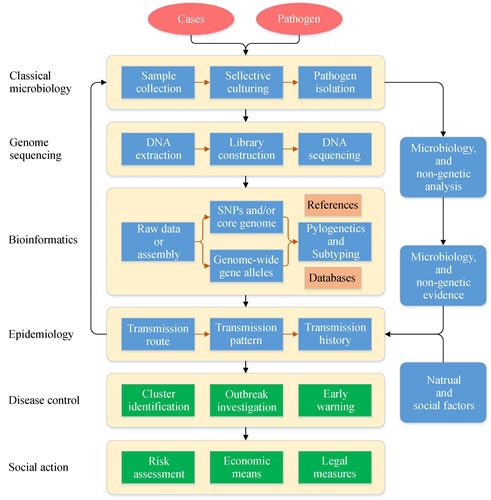
自从利用新的测序策略获得第一株细菌全基因组,至今已过去二十年,这期间,基因组测序和信息分析技术仍展现出了快速的发展,测序方法和序列读长都在迅速改善,原始测序与组装质量、序列数据挖掘分析策略和算法不断优化。通过微生物基因组序列比对分析,能够准确获得变异特征,这些序列变异特征可作为识别不同亚型的生物标记,并据此分析不同菌株间的遗传变异关系,为数字化重构菌株间的流行病学关联程度建立了分子基础。基因组信息在传染病监测和暴发调查研究中的应用可被定义为:根据基因组信息分析病原菌分离株之间的分子流行病学关系,结合社会信息、自然与地理信息等,重构传播链,协助分析解释传染病的发生和扩散,对疫情进行溯源,进而明确传染病的发生模式(图1)。目前分离株基因组比对主要有两种方法,分别是基于序列的全基因组/核心基因组单核苷酸多态性(SNP)、基于来自全基因组/核心基因组的多位点序列分型(MLST)。利用病原细菌分离株的基因组序列,可在下述几个方面发挥作用。
图1 细菌性传染病暴发调查和监测分析中基因组流行病学研究的一般流程。
1. 鉴别暴发
在日常监测中,对表现为散发的病例分离株,通过基因组测序比对,能够从其中发现一些序列一致或非常相似的菌株,提示这些病例可能有共同/连续暴露,感染了同一来源或相关联来源的病原细菌。这些信息可反馈给流行病学调查人员启动调查,就可能会发现暴发。这能够在一起暴发疫情的早期就发现共同暴露的病例,通过控制措施避免疫情扩大,从而发挥预警和早期控制的作用。用这些菌株序列比对不同食品、环境、媒介等来源的菌株序列,能够精确地发现或证实感染来源。这种策略和方法已用于多起疫情比如单增李斯特菌、耐甲氧西林金黄色葡萄球菌等的感染调查,给出了精准的结果。全基因组测序在常规监测中鉴别暴发的一个关键是确定多少遗传差异以内可以判断为流行相关的簇,需要根据各个菌种的进化特点、分子钟、流行病学特征来建立综合的判断标准。
2. 追踪病原菌传播
全基因组比对分析技术能够针对一起暴发疫情追踪到病原菌的精细传播链。基于细菌基因组的传染病暴发溯源方法在跨洲、跨国、地区局部暴发、院内感染暴发的调查中均已起到很好的作用。2011年海地霍乱疫情调查是基因组数据应用于追踪病原菌传播的经典案例,其进步之处在于基因组数据区分出PFGE不能区分的菌株,发现跨洲传播的病原证据,该克隆长期存在于东南亚、并经尼泊尔扩散到海地。国内比较早的应用是针对2010年云南甲型副伤寒暴发的调查,基于分子分型、全基因组序列比对及流行病学调查,发现暴发有一个主要型别在县城扩散、源头是含有甲型副伤寒沙门菌污水所污染的菜地中所生长的蔬菜。
3. 发现新的传播模式和途径
获得病原细菌分离株的全基因组序列,能够更精细地分析病原菌变异特征,实现精准区分克隆群和传播关联的作用,因此,在准确发现传播模式和分析长期动态变化等方面,全基因组数据相比于传统分子分型方法更加有效。
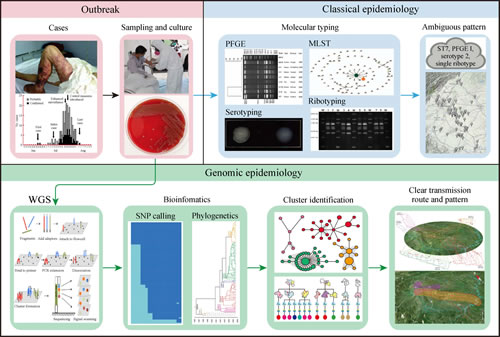
全基因组分型溯源技术在中国四川省2005年发生的人感染猪链球菌病暴发调查中发现了比以前分析更多的病原体遗传变异和流行病学特征(图2)。以往的流行病学调查、实验室检测和分子分型只能明确携带猪链球菌或发病的猪为传染源,患者通过与猪的直接接触受到感染。通过基因组测序和分析揭示了病例是在各自区域内受到感染的,为多点平行传播模式,这也得到其他信息的支持,包括当地生猪的养殖模式、地理、交通和经济因素等。另外一项对于中国副伤寒沙门菌多发地区传播特征的研究,利用基因组信息揭示中国菌株传播呈现两个模式:由沿海省份传入内陆多发省份、以及在多发省份各自循环扩散。这些发现也得到了人口流动和经济转型等社会学数据的支持。自然和社会环境的不同或改变、人类活动等因素均能导致传染病传播模式的不同,而应用基因组流行病学结合社会经济、自然地理等因素,可以深入认识这些新型的传染病传播模式。
图2 全基因组分型溯源技术对2005年中国四川人感染猪链球菌病暴发中识别出了比以往分析(上半图)的新的认识(下半图),病例在各自区域内受到感染,为多点平行传播。
4. 发现新的克隆群
新出现的流行克隆群和暴发菌株会呈现一些新的基因组特征,例如新的毒力基因、环境适应基因、耐药基因等,这些表型特征能在流行或暴发中导致高病死率和治疗失败。另外这些新基因组标识也可用来确定新克隆群的标记。例如在2010年海地霍乱暴发、2011年欧洲O104:H4大肠杆菌暴发中,研究人员均通过基因组测序和分析确定了暴发菌株的毒力基因特征,这些特征随后被作为追溯来源的标记之一,以及解释高致病原因。在猪链球菌中,通过基因组序列区分出一些高致病的潜力的、具有公共卫生意义的克隆群。在嗜肺军团菌中也区分出具有细胞内低存活能力的遗传克隆,并且反推出一些具有潜在细胞内存活和重要致病意义的基因。
5. 病原细菌基因组学监测的网络化
由于传染病能够跨地区传播,因此一方面不同地区流行的传染病在病原学上各有特征,另一方面具有共同遗传特征的暴发株可在不同地区出现,因此需要开展以病原菌基因组测序比对为技术基础的、不同地区共同参与的网络化实验室监测。目前美国的FDA、英国公共卫生署、国际病原细菌分子分型监测网络PulseNet、全球微生物识别组织(GMI)以及中国的国家致病菌识别网均在探索和应用全基因组测序分析技术来开展细菌性传染病监测。这个网络需要标准化方法、信息化平台,使数据分析和监测应用达到实时与迅速的网络传播。
展望
目前的分析依然要求获得纯培养的细菌,从标本中开展宏基因组测序及组装识别出病原菌更长的特征序列、以及单细菌测序,是将来病原细菌基因组学技术的攻关目标,使在疫情监测中更快获得病原菌的基因组数据。病原细菌全基因组测序质量和大数据分析能力需要进一步优化,并整合多种监测数据形成病例流行病学分析大数据、应用于传染病暴发识别预警的判别。需要更多的国家和地区建立基因组数据库和查询网络,从而进一步形成全球性的网络,识别不同特征的菌株、发现聚集性病例、预警和溯源暴发。(来源:科学网)
微信号:frontmed
特别声明:本文转载仅仅是出于传播信息的需要,并不意味着代表本网站观点或证实其内容的真实性;如其他媒体、网站或个人从本网站转载使用,须保留本网站注明的“来源”,并自负版权等法律责任;作者如果不希望被转载或者联系转载稿费等事宜,请与我们接洽。